SWING
SWING, Small and Wide angle X-ray scattering
Pour l'étude d'échantillons à structure complexe : macromolécules, nanomatériaux, tissus...
En fournissant des informations sur la structure de la matière à des échelles variant entre le nanomètre et le micromètre, la ligne de lumière SWING permet de répondre à de nombreuses questions liées à la matière molle, condensée, à la conformation des macro-molécules en solution (BioSAXS) et aux sciences des matériaux.
La configuration de la ligne permet la mesure simultanée des rayons X aux petits angles (SAXS) et aux grands angles (WAXS) dans une plage d'énergie allant 5 à 16 keV. Des expériences de diffusion anormale peuvent également être effectuées.
Une très grande variété de types d'échantillons peut être étudiée, par exemple, des solutions, des gels, des solides amorphes, des solides cristallisés, tout cela grâce à la diversité des environnements d'échantillons proposés.
Flyers


L'équipe


* Prestataire extérieur, intérimaire ou collaborateur
Les actualités
Toutes les actualités de SWING
École européenne HERCULES 2026 à SOLEIL
Production d’hydrogène - un électrocatalyseur prometteur basé sur des nanotubes d’argile
Les vidéos
Toutes les vidéos de SWING
Les lumières de SOLEIL (VFSTF et LSF) (2/3)

SWING : Etude des protéines par chromatographie
-
Données techniques
- Source
-
In-vacuum U20 undulator.
Source Size (sigma, μm): 388 (H) x 8.1 (V)
Source Divergence (sigma, μrad): 14.5 (H) x 4.6 (V)
- Gamme d'énergie
-
Between ~5 and 16 keV
- Résolution en énergie
-
~2 eV
- Optiques
-
Diaphragm at 11.7 m (1x0.5 mm2)
Fixed exit DCM Si111at 20 m
Fixed incidence focusing KB at 22.5 m (HFM removable for micro-focus)
Linear H-CRL (f=81 cm): 31 m
Sample position: around 32 m
Detector / Sample Distance: 0.5 – 6.5 m
- Taille du faisceau
-
200 x 20 to 500 x 200 μm2 (KB)
20 x 20 µm2 (micro-focus)
- Flux sur l'échantillon
-
(with 500 mA ring)
1.1013ph/s @7keV,
5.1012ph/s @12keV,
1.1012 ph/s @16keV
- Beamstops
-
Two active beam stops are available, including:
• a Si diode (Hamamatsu); external size 6 x 3 mm², for the µ-beam setup.
• a Diamond diode, external size 2.2 x 1.2 mm², for the standard SAXS setup.
- Détecteurs
-
SAXS:
• EigerX4M in vacuum (Dectris web site)
• Detection area: 162.5 x 155.2 mm2
• pixel size : 75 x 75 µm2
• frame rate : up to 750 Hz
The Eiger is positioned on a 3 axis translation stage, allowing large vertical and horizontal repositioning with respect to the beam. The third translation is to change the Sample-Detector distance.
WAXS:
• Merlin (Quantum Detector website)
• Detection area: 28x28 mm2
• pixel size: 55 x 55 µm2
• frame rate: 1000 Hz
The detector is positioned in air on a double axis goniometer (Mecaconcept Company), with a further three translation stages for detector alignment with respect to the sample. One more translation along the X-ray beam direction enables to set the sample to detector distance. Rotation of the detector stage around the beam (χ axis) gives the possibility to explore anisotropies at fixed q value.
- Q range
-
All values below suppose that the direct beam is close to a detector corner.
q Max values are on the opposed corner.
q Min values are on the vertical direction only.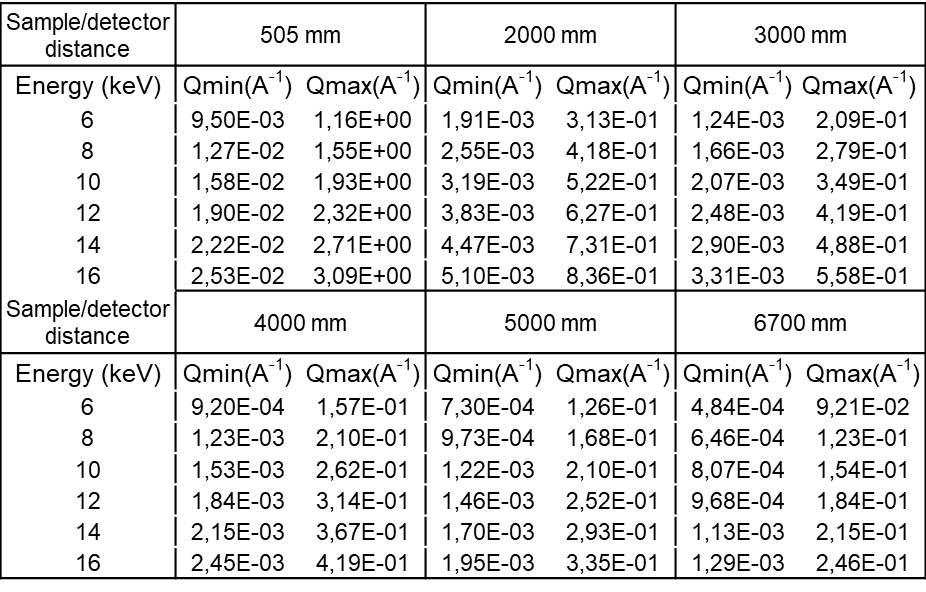
- Motorized Stages for Sample Environments
-
TX1/TZ1, the large main motorized stage, can be used to bear heavy equipments, as it is or in combination with load bearing tables. A more accurate motorized table, TX4/TZ4, can be used on top of TX1/TZ1 for lighter equipment.

Dedicated sample environments have been developed to provide scientific communities with optimized experimental conditions. For soft-condensed matter and material science, a number of devices are available. For structural biology, a completely automated BioSAXS system is routinely used including either online purification or direct injection, connected to a quartz capillary cell placed within a vacuum chamber. In addition, don’t hesitate to ask for help in the optimization of your own sample environment.
All sample environments, except the rheometer, are preceded with an On-Axis-Visualization (OAV), for direct visualization of the sample.
Here are presented most of the devices proposed on the beamline.
BioSaxs: HPLC-SEC
 | • Sample volume: 10 to 100µL • Sample Concentration: 1 to 30mg/mL • Two parallel purification circuits (two columns with different buffers can be inserted) • Connected to the Biosaxs Vacuum Cell • Biosec-3 columns available on the beamline • Combination of the SEC-SAXS system with MALS and RI at user request • Automated workflows for sequential multiple injections and data analysis • IspyB database |
BioSaxs: AutoSampler
| • Sample volume: 10 to 50µL • Sample Concentration: 0.2 to 10mg/mL • Duty cycle: 3.5 min (pipetting, injection, measurement, cleaning and drying) • Vials capacity: 104 • Thermalized vials holder (5-10 to 60 °C) • Injection rates of : 10 to 100 µl.min-1 • Viscosity compatible up to 30 % glycerol in water / 250 g.l-1 BSA • Connected to the Biosaxs Vacuum Cell • Immediate switch between direct injection and SEC-SAXS modes |

BioSaxs: Vacuum cell
|

High Pressure for Liquids
|
Ex: from 1 to 3000 bars and back: 35min in 20 steps |

Static Capillaries Holder
| • Horizontal or Vertical orientation • Number of slots: 20 capillaries (advised: Ø 1,5 mm) • Scanning window behind each capillary: 5x1.2 mm2 • Distance between capillaries: 5 mm • Thermalization: 5-60 °C |

Flow-through Capillaries Holder
| • Horizontal Ø 1,5 mm open quartz capillaries: 3 • horizontal scanning window: 20 mm • distance between capillaries: 15 mm • Thermalization: 5-60 °C Circulation through the capillaries can be done using various setups : • Peristaltic pump • Chemsaxs Injection Robot • 2-Syringe motorized driver • Manual injection |

Gels / Solids (polymer slice, pellet) Holder
| • Two rows: 2 x 20 wells • Distance between rows: 45 mm (V) • Distance between wells : 15 mm (H) • Wells diameter: 2 and 4 mm • Teflon spacer thickness: 1.5 mm • Closed on each side by a Kapton foil
Warning : this gel holder won’t work with low viscosity gels (leaks) |

Liquids / Gels holder
| • 16 sockets for low viscosity gels and liquids • Stainless Steel spacer path: 1.5 mm ( 1.7 mL) or 1 mm (1.1 mL) • Closed by 2 mica windows
Tip: Sealing was tested with ethanol. Sealing was efficient for more than a week. |

Mixing and pipetting cell
| • 5 / 10 ml mixing cell with a steerer • Sample aspiration through a Ø1Ø 1,5 mm quartz capillary • Possible injection into the mixing cell through a separated hole, synchronized with detection • Thermalization 5- 60 °C Tip: The sample is drawn in the capillary at the required height. After X-ray exposition, it is reinjected into the mixing cell. |

Linkam THMS 600
|
|

Traction Cell
| • Max. Working load: 111 N • Max. working length : 6 mm • Resolution 0.05 N • Step : 0,5 µm |

Rheometer
| Rheometer Anton Paar MCR 501 • Polycarbonate Couette cells • Internal radius: 10 mm, • Height : 17 mm • gaps (Re – Ri): 0.5 / 0.2 mm • Sample Volume: about 1.2 ml • Tangential and radial positions for x-ray beam |

Stop-Flow-SAXS-MALLS
|
• Tip: Please contact us for double connection to MALLS and SAXS
|

Micro-fluidic
| • Fluigent MFCS-EZ system • 3 pumps available see section: specific setup/microfluidics • 3 flowmeters see section: specific setup/microfluidics • Maximum injection pressure for each pump: 345 mBar
Monitoring of the commercial pumps and flowmeters is decoupled from the beamline workflow and is thus controlled from a separate laptop. |

ChemSaxs Liquid AutoSampler
|
|

StopFlow-MALS-SAXS
| • Biologic SFM400 • BioSAXS Vacuum Cell • MALS: Wyatt 18 angles Heleos-II • Total volume per injection : 1200 µL • Thermalized tubings • Automated cleaning (background buffer, cleaning buffer, detergent, water) • Synchronization between SF, SAXS and MALS acquisition via TTL signals • Mixed solution continuously refreshed in the SAXS cell • Dedicated GUI for cleaning and acquisition launching Example of what can be obtained with this facility (2.29 MB) Please contact us for this sample environnement |

User’s environment
Your own setup is welcome on the beamline. Please contact us before submitting a project, as we need to be sure that your setup can be installed on the beamline.
It is possible to insert the control of your setup in the data collection workflow
- Remote control of your monitoring laptop
- Send to or receive a trig from your setup
- This allow synchronization with the data collection workflow
Under one of the two specific conditions:
- A tango device (software) has already been developed for that apparatus. Unlikely.
- That apparatus includes a connection for external triggering (in/out, BNC preferred), that we can connect to one of our signal generating or reading electronic cards . Likely . (mainly 5V; 0 to 24 v possible; with BNC type cables).
Specific Setups
Linkam THMS 600
This apparatus requires your capillaries to be 1.4 mm in diameter maximum. As the Linkam “oven” part is really small and its environment crowded, those capillaries need to be cut-off and sealed-off. For small diameter capillaries, we recommend to center them on the incoming beam aperture by inserting a (homemade) shim.
To fit in the Linkam oven, it is mandatory to cut your capillaries to a maximum length of 35 mm. In order to obtain a clean cut, quartz capillaries are preferred to borosilicate.
You can either cut your capillaries in your lab or at SWING. We can show you how to do it.
After the sample is loaded in the cut capillary, you have to seal it in order to avoid evaporation of solvent or spillage in the Linkam stage. Two techniques can be used:
Flame sealing if you have borosilicate capillaries. If you have a glassware lab you still can seal quartz capillaries with your glassmaker.
Glue sealing for Quartz capillaries
In this case, we cannot firmly recommend any glue as it is sample and temperature dependent… but epoxy glues are of good use.
If glue sealing does not suit your requirement, you can give it a try with modeling clay (if you don’t heat over 120 °C)
Microfluidics
SWING is equipped with:
- A MFCS-EZ system (Fluigent) with 3 pumps installed. The maximum pressure each pump can give is 345 mBar.
- A flowrate platform, consisting of 3 flow unit models and the flowboard.
Please find the flow unit characteristics:
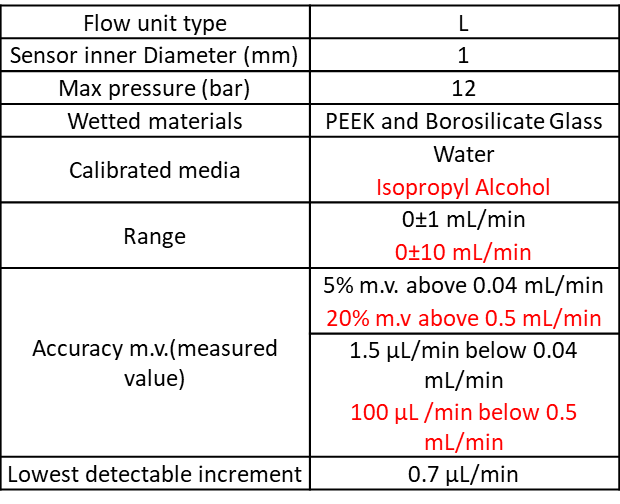
This setup can use either three 5 mL Eppendorf and or one 15 and/or three 50mL Falcon and or one for classic chemistry glass bottles (GL45 screw cap).
This system is controlled via Maesflow, the proprietary software. It allows:
- Direct control on the pump pressure = no active control on the flow rate. But flowrate value readable.
- Direct control on the flow rate = active control of the flow, keeping each flow stable vs the other flow.
There is no feedback from the software or microfluidics controller to the beamline control system. So you will have to control both systems from the control room.
As microfluidic chips are designed for a specific experiment, SWING staff does not provide chips. Nevertheless, if you need to make your chips at SOLEIL, we will be pleased to welcome you to the Microfluidics Lab. Furthermore, we will be happy to help designing your chips: choice of materials, size of channels for saxs measurement…
Please contact us in advance as the sample environment may be crowded and thus your chips, tubbing and other device may not fit easily in the beam path.
Microfocus and On-the-fly Raster Scan
Microfocus and On-the-fly Raster Scan can either be used as a combination or independently.
Microfocus
Installation of the microfocus setup requires more time than for a usual sample environment. Thus it is mainly installed for a whole week.
SWING Microfocused beam is obtained using a linear H-CRL for horizontal focusing and the standard VFM (vertical focusing mirror) for vertical focusing. The resulting beam size is of 20 x 20 µm² on the sample.
With this setup, the sample environment is much more crowded than for a “classic SAXS” experiment. The TX4/TZ4 table can be used to move samples holders. If you want to use your own holder please contact us to know if it will fit.

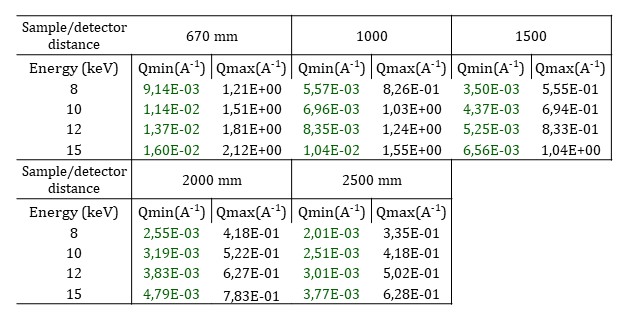
As the beam is strongly focused on the sample, the sample to detector distance is limited from 0.670 to 2.5 m and a larger beamstop than the usual one is positioned. Moreover, the number of selectable H-CRLs limits the working energy to any of four fixed values: 8, 10, 12, and 15 keV. The resulting q-ranges are shown in the following table.
Raster scan
Continuous 2D imaging is called “Flyscan” at SOLEIL, by opposition to step-by-step scan. It consists of a 2D cartography whose first dimension is associated with a continuously moving motor and the second dimension is a standard step movement.
Flyscan is mainly used with the combination of the Microfocused beam. Yet it is still possible to use Flyscan with a more standard beam.
In Flyscan mode, the data from the sensors (detectors, diodes, thermocouple…) and motor positions are all collected synchronously and merged in the same HDF5 file. This allows reconstructing maps from each of the scalar sensors or from an operation made on the images.
For a map of 200 by 200 pixels, with a typical exposure time of 200 ms (plus dead time of 5ms), motor steps of 25 µm, the scan will last about 1 hour and a half, about 6 times faster than the equivalent step-mode scan. (voir presentation sur le overhead)
As any for any session at SWING a specific data collection Passerelle GUI and a specific workflow visualization COOX GUI are available.
At the moment, the generation of maps using Foxtrot is under development, it will be available on the beamline in September 2020.
Before submitting your project
Don’t hesitate to contact one of the beamline scientists, for any information about:
- The Beamline
- The available sample environments describe above
- Bringing your own sample environment
- Your project feasibility
- How many shift your project requires
Aurelien Thureau (aurelien.thureau@synchrotron-soleil.fr) and Javier Pérez (javier.perez@synchrotron-soleil.fr) deal with Biosaxs
Thomas Bizien (thomas.bizien@synchroton-soleil.fr) and Javier Pérez (javier.perez@synchrotron-soleil.fr) deal with “everything else”
Useful links:
- Application for beamtime:
https://www.synchrotron-soleil.fr/en/users/application-beamtime
- Project submission on the sunset:
https://www.synchrotron-soleil.fr/en/users/application-beamtime/how-prepare-proposal
- Participant declarations and sample declarations:
https://www.synchrotron-soleil.fr/en/users/preparing-your-experiment-administrative
Tip: Be careful, don’t submit before your sample and participants lists are complete. Bear in mind that after selecting samples and participants on the sunset interface, you can “save only” rather than “save and submit”. Thus if your selection is incomplete, you can still edit your previous list and submit it in time when you have your final list ready, as it is requested here: https://www.synchrotron-soleil.fr/en/users/preparing-your-experiment-scientific/safety-requirements#SamplesSubstances
Support labs:
https://www.synchrotron-soleil.fr/en/know-how/supports
Tip: Biolab1 and Chemistry 1 are close to the SWING beamline.
For any complementary information,
Please contact the User Office:
+33 1 69 35 96 51
Working hours are (from Monday to Friday):
8:30 - 17:30
Office A1.0.06, Main Building
Coming at Swing
Your session generally includes some time needed for set up installation. It is strongly recommended to discuss with your local contact before coming to the beamline to save time during installation. In particular, don’t hesitate to ask for help in the optimization of your own sample environment.
Please bring an external drive to back up your data from SOLEIL mainframe (at least 1 TB). It has to be compatible with Windows format (FAT32, exFAT or NTFS). Your local contact will run a macro for live synchronization between data on SOLEIL mainframe and data on your hard drive. However, even if we do not advise to do so, you can still download your data files a posteriori, via the sunset [Experiment Data][Soleil Data Retreaval]
Wi-Fi is available on SOLEIL site using your sunset login and password or using Eduroam network.
BioSaxs
What you have to bring
We use Agilent standard vials and inserts for the HPLC device and the Auto Sampler. We lend you the vials and the caps but you have to buy your own inserts (Agilent reference: 5181-1270). In case you forgot, we can provide you with used inserts that you can wash and dry.
The minimum quantity of buffer is 40ml for one SEC-SAXS experiment (10mL of system purge + 20 mL of column equilibration + 10mL of elution). We recommend you to bring much more (100mL to 500mL) to obtain a better equilibration of the column and to reduce stress in case of trouble.
What we can lend
The biochemistry preparation laboratory provides support to users for the preparation of their experiments. For any question, please ask the laboratory staff.
You are free to bring your own analytical columns for SEC-SAXS. We have also a wide selection of columns that we can lend to our users. All these columns have been calibrated:
BioSec 2.7-130 - Users25 pH: 2 - 8.5 Size: 0.1 - 100 kDa | 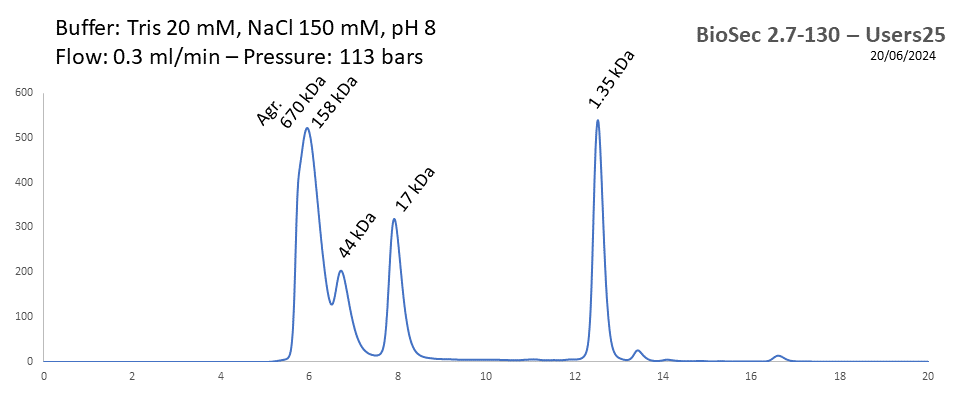
|
BioResolv 2.5-200 - Users29 pH: 2 - 8 Size: 10 - 450 kDa | 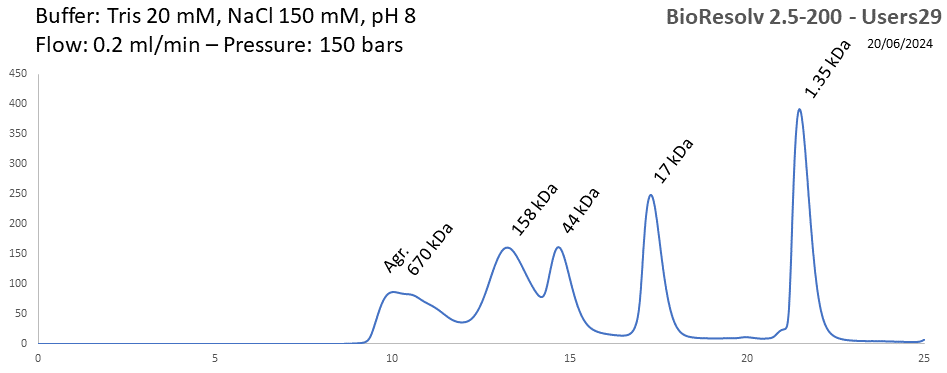
|
BioSEC 2.7-300 - Staff15 pH: 2 - 8.5 Size: 5 - 1250 kDa | 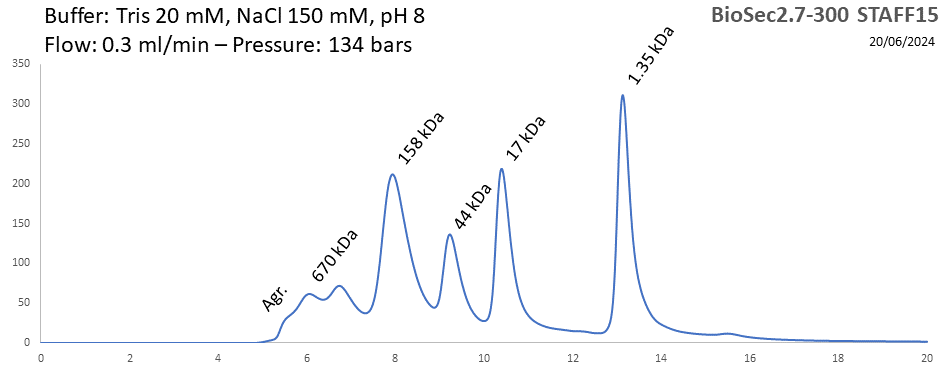
|
BioSEC 2.7-300 - Users31 pH: 2 - 8.5 Size: 5 - 1250 kDa | 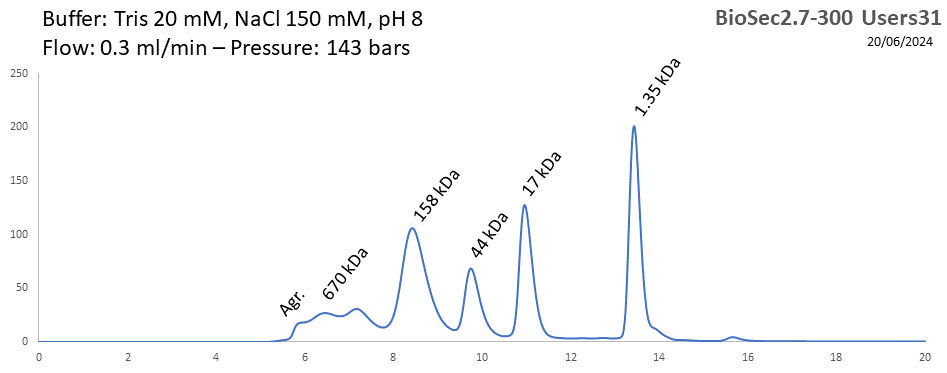
|
BioSEC 3-300 - Users33 pH: 2 - 8.5 Size: 5 - 1250 kDa | 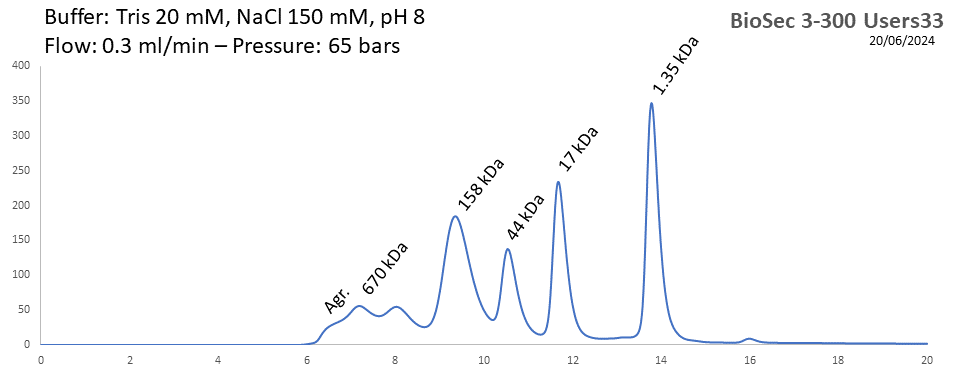
|
BioSEC 3-300 - Users32 pH: 2 - 8.5 Size: 5 - 1250 kDa | 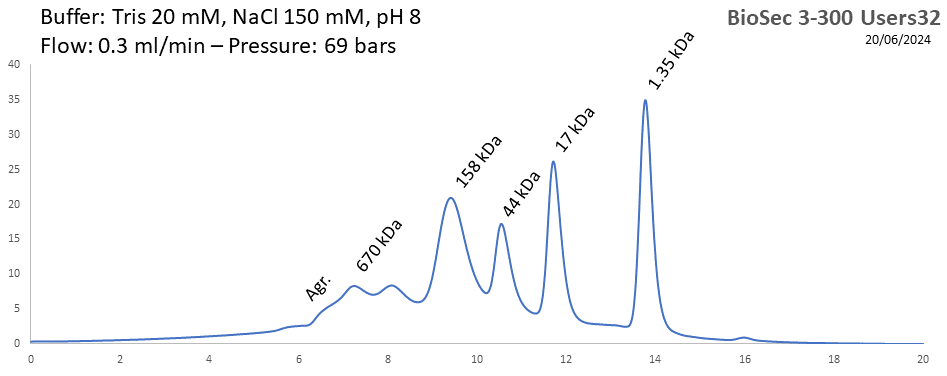
|
BioSEC 5-500 - Users28 pH: 2 - 8.5 Size: 15 - 2000 kDa | 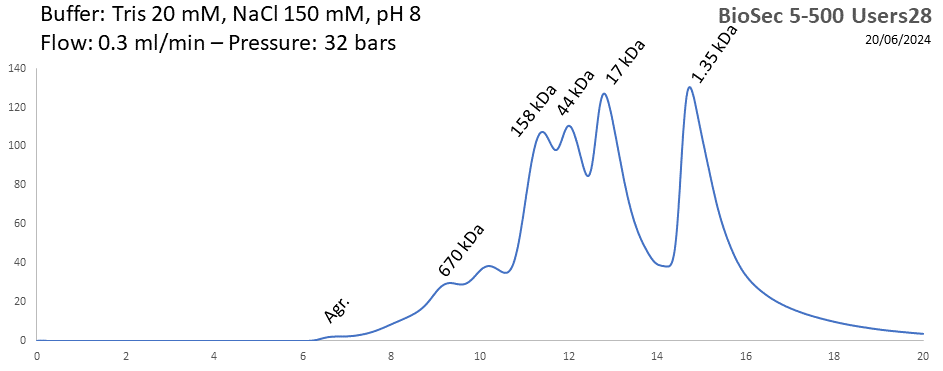
|
BioSEC 5-1000 - Users34 pH: 2 - 8.5 Size: 50 - 7500 kDa | 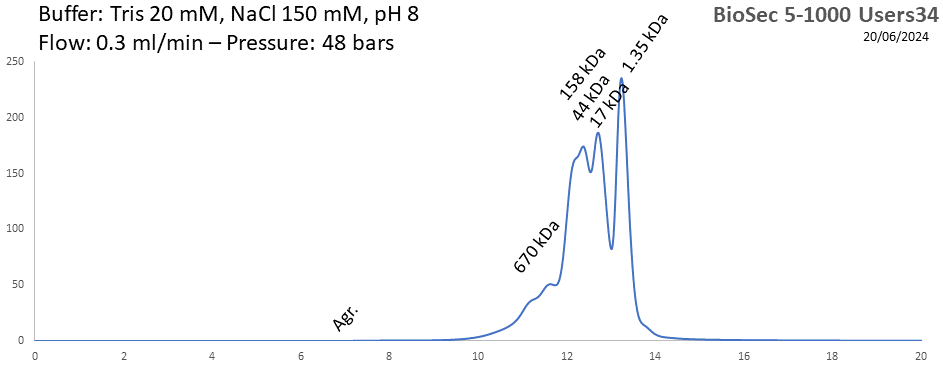
|
ChemSaxs
What you have to bring
You have to bring your own capillaries (Ø 1.5 mm preferred) and specifics tools if required for your own setup, or for preparing your samples. We use Agilent standard vials and inserts for the ChemSaxs Liquid AutoSampler. We lend the vials but you have to buy your own inserts (Agilent reference: 5181-1270). In case you forgot, we can provide you with used inserts that you can wash and dry.
Mail-in services
Mail-in services are only proposed for accepted and planified proposals, in the case where users are not able to come onsite.
A discussion between the user and the local contact must be conducted beforehands to agree on the experiment conditions.
Mail-in experiments are conducted by staff member and a few restrictions apply :
- The sample preparation onsite should be as simple as possible, if any.
- Experiments will be carried out only during working hours (8h00 to 20h00)
- An acquisition sequence might be launched outside working hours but without monitoring of the local contact
- Users are strongly suggested to follow the experiment using the remote access tools offered by SOLEIL
For instructions the
biosaxs mail-in document for usersneed to be followed. A
samplesheet BioSAXSmust be filled and sent as soon as possible before the experiment date.
During session
Keep in mind that your session can include beamline setup and sample environment installation.
The local contact will take the necessary time to make you completely autonomous on the beamline.
Data Collection sequences
All data collection sequences appear to the user as a simplified GUI with a list of parameter fields to fill (based on Passerelle workflow language). For most of the sample environments and “classical” data collections, a sequence is already written.
If necessary, a new collection strategy can be written just before your experiment, with a new GUI automatically generated. To do so, don’t hesitate to ask your local contact.
It is possible to include an action on an external apparatus (that you bring to SOLEIL) to the data collection workflow under one of the two specific conditions:
A tango device (software) has already been developed for that apparatus. Unlikely
That apparatus includes a connection for external triggering (in/out, BNC preferred), that we can connect to one of our signal generating or reading electronic cards.
Monitoring
Specific GUIs (based on COOX platform) give the possibility to monitor the beamline state (vacuum, valves, motors positions and states…) and the acquisition progress (live images from detector, live values from diodes, direct visualization of the sample,…) as well.
Simple actions like changing sample to detector distance, closing/opening the safety shutter, realigning the monochromator rocking curve, changing the working energy, …, can be done directly from the COOX GUI by clicking on active buttons.
Data Reduction
All data produced on the Swing beamline are stored in HDF5 files. Those files contain detector images, diode values, sample pictures, and a lot of beamline metadata. The files can be processed by the Java based self-installing software Foxtrot. It allows, among other functions: Data visualization (2D images from detectors, Transmission curves, …), data reduction (from 2D images to 1D curves, normalization included, gives intensity scale in cm-1, …), mathematical operations (curves averaging, curves subtraction, curves merging, useful if you recorded data at 2 or 3 sample to detector distances, other basic functions :scaling, linear function, division, summation, …), fits (Guinier, Gaussian and Lorentzian peaks, polynomial).
You will be trained by your local contact to use Foxtrot. You will be provided with a copy of the software as a zip file, including your masks and reduction macros.
For SEC-SAXS and BioSaxs AutoSampler, an automatic workflow for data analysis is available. This workflow takes raw images as input files and sends all results curves into the IspyB database. All intermediates files are available to users and can be reloaded into the Foxtrot application.
Living Area
We have a living area with a self service although not free ;-), coffee machine,a sofa a fridge for food and beverages, a micro wave and a table.
This area is sample freee, Please clean it after your session.
After your session
After your session
- Sign the Safety Authorization Sheet (SAS).
- Please clean the living area and the experimental hutch. If you used support labs, please leave them clean.
- Please don’t forget to fill the end- of- run report in the sunset webpage.
- It takes 5 minutes ! That is the best way to specify your remarks.
- These reports are read by many people at Soleil, including sometimes the management.
Préparation des échantillons et acquisition
Tutoriels Foxtrot
Présentation des BioLab 1 & 2
Listes des logiciels développés par la ligne SWING.
Programme Foxtrot
Foxtrot Software (Data reduction and treatment) can be downloaded here
Your mask(s) and macro(s) will be given to you by your local contact during the session.
Programme Memprot
Programme Denfert
Programme Denfert (Détermination ab initio d'enveloppes moléculaires, incluant une couche d'hydratation)
version 2.0 (June 2015)
Denfert : F.A.Q.
A zip archive containing executables for Mac, Linux and Windows together with a test example may be downloaded by clicking here.
... a program for the ab initio “dummy-atom” structural modeling of Biological Macromolecules including the contribution of their inherent hydration layer.
Developed by Alexandros Koutsioubas and Javier Pérez
If you use DENFERT, please cite :
A. Koutsioubas & J. Pérez, Journal of Applied Crystallography (2013) 46, 1884 and A. Koutsioubas, S. Jaksch & J.Pérez, J. (2016). J. Appl. Cryst. 49 (doi:10.1107/S1600576716003393)
Brief Description
DENFERT is implementing a simulated annealing algorithm similar to DAMMIN program by D. Svergun (Biophys. J. 76, 2879-2886) for the restoration of low-resolution structural info of bio-molecules from SAXS and SANS data. The major advantage of DENFERT is that the hydration layer around bio-molecules is taken into account by introducing a second type of beads (hydration beads) in the model.
In the top figure, we see an example of the shape restoration of Lysozyme from SAXS data using DENFERT. A cartoon representation of the crystallographic structure is also presented for comparison. The bottom figure depicts additionally the hydration layer around the reconstructed protein shape.
denfert manual (126.1 Ko)
Dadimodo
DADIMODO is a program for refining atomic models of multidomain proteins or complexes against small-angle X-ray scattering data. Domain structures are mainly kept rigid and can be user defined. Stepwise generic conformational changes are applied cyclically in a stochastic optimization algorithm that performs a search in the protein conformation space. The algorithmic structure guarantees that a physically acceptable full atomic model of the structure is present at all stages of the optimization.
To prepare your complete pdb files, you can use our![]() pre dadimodo script (3.71 Ko)(remove the _.txt extension).
pre dadimodo script (3.71 Ko)(remove the _.txt extension).
Downloads
BioSaxs course (8.07 Mo)