Une nouvelle méthode d’identification des structures secondaires et du repliement des protéines
L’analyse de la structure de fibrilles d’amyloïdes de différentes protéines impliquées dans des pathologies comme Alzeihmer, Parkinson ou le syndrome d’Huntington représente un réel défi. Il a fallu les efforts conjoints du Groupe de Recherche en Neuro-immunologie (Académie des Sciences et Eötvös Loránd University Budapest, Hongrie), de l’Institute for Protein Research (Université D’Osaka au Japon), et de la ligne de lumière DISCO du synchrotron SOLEIL pour caractériser ces fibrilles d’amyloïdes jusqu’à leur organisation spatiale dans des conditions physiologiques. L’équipe internationale a mis au point un nouvel algorithme et une approche spectroscopique innovante, s’appuyant sur le dichroïsme circulaire utilisant le rayonnement synchrotron (SRCD), qui rendent enfin possible la résolution de la structure de ces agrégats de protéines.
La spectroscopie par dichroïsme circulaire (CD) repose sur une absorption différenciée de la lumière selon qu’elle est polarisée circulairement droite ou gauche. Les chromophores biologiques, comme les protéines par exemple, ont des spectres d’absorption CD dans la région des ultraviolets lointains distincts et qui dépendent de leur structure. Les sources de lumière synchrotron, reconnues pour leur grande intensité lumineuse et une plus grande gamme spectrale disponible que les sources conventionnelles, ont fourni aux biophysiciens des spectres plus précis, collectés en un temps plus court, mais aussi une quantité d’informations plus importante.
L’analyse minutieuse de la torsion des feuillets beta, éléments de structure secondaire des protéines, corrélée avec des formes de spectres caractéristiques obtenues par CD dans la région des UV lointains a permis de différencier les feuillets beta parallèles et antiparallèles.
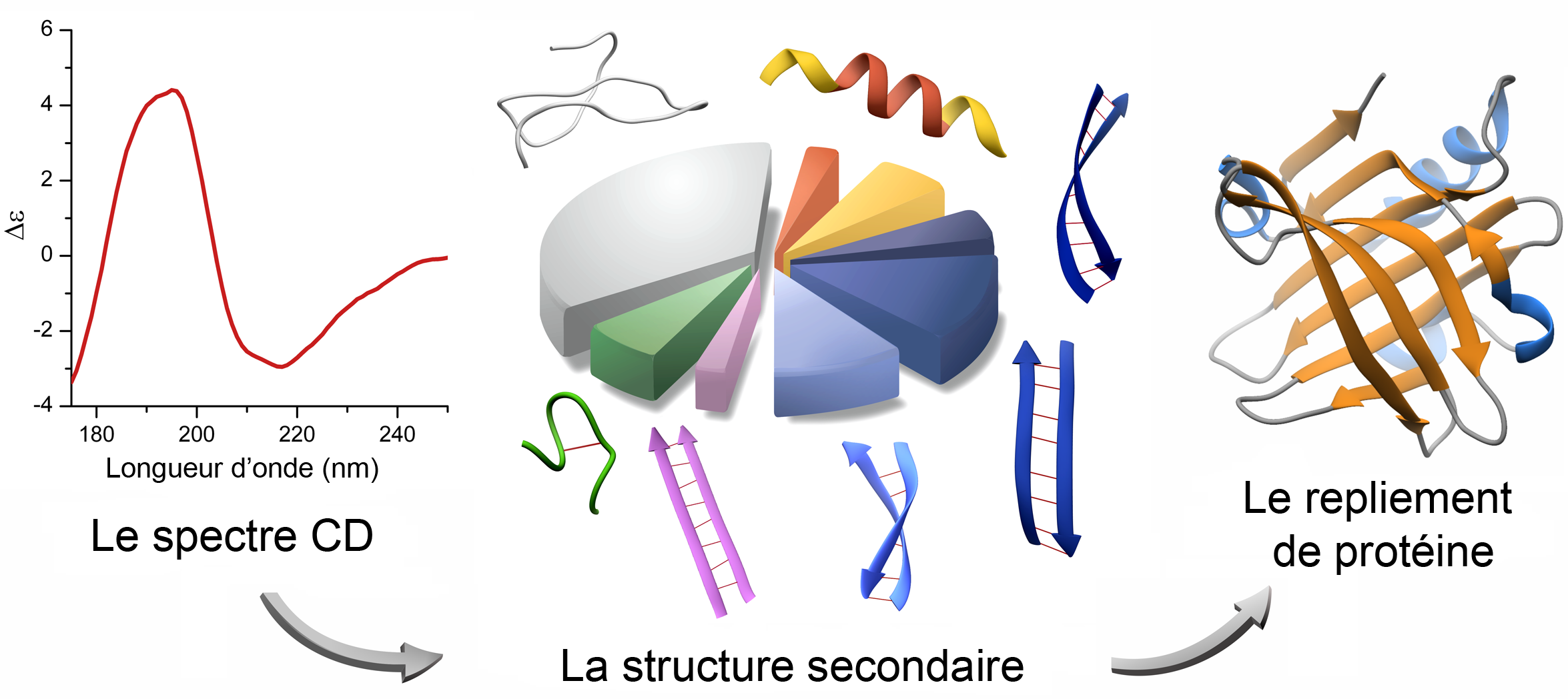
Les informations structurales supplémentaires ainsi obtenues ont mené à l’identification précise de la structure secondaire, permettant ainsi de prédire le repliement des protéines. Le repliement des protéines est alors classé en fonction du schéma spécifique des éléments de structure secondaire. L’ensemble de la méthode a été intégrée dans un nouvel algorithme bio-informatique (BeStSel). Par ailleurs, outre les structures riches en feuillets beta, la méthode peut s’appliquer avec une excellente précision à n’importe quelle protéine. Les chercheurs espèrent ainsi aider la communauté scientifique en lui fournissant un outil puissant pour l’analyse rapide et fiable de structure, ce qui est particulièrement utile lorsque les techniques utilisant les rayons X ou de Résonance Magnétique Nucléaire (RMN) échouent.