Profiler l’interface liquide-vapeur à l’échelle de l’angström
A quoi ressemblent les tout premiers nanomètres de l’interface entre un liquide et le gaz environnant, au niveau moléculaire ? La question est loin d’être triviale, et les techniques permettant expérimentalement de faire un « profilage » de la densité des différentes espèces atomiques en fonction de la distance à l’interface sont rares.
Pour y répondre, une collaboration de scientifiques du Fritz-Haber-Institut (Berlin) présente une nouvelle méthode, basée sur la mesure de l’angle d’émission de photoélectrons dans un microjet liquide irradié par le rayonnement X de la ligne PLEIADES. Des différences de profondeur moyenne de seulement 1 Å (10-10m) environ ont été mises en évidence entre différentes classes d’atomes.
L’interface entre les phases liquide et gazeuse joue un rôle important dans de très nombreux domaines, par exemple en chimie atmosphérique où de nombreux processus physico-chimiques ont lieu à l’interface entre les aérosols liquides, ou entre l’océan et l’atmosphère. Caractériser expérimentalement ce type d’interface au niveau moléculaire, dans les quelques nanomètres qui séparent les deux milieux, reste difficile. D’importantes avancées ont été possibles ces 20 dernières années grâce au développement de la spectroscopie aux rayons X de photoélectrons (XPS) sur les liquides. La XPS classique est une technique sondant la surface des échantillons et permettant de déterminer les espèces atomiques présentes et leur environnement chimique. Elle repose sur la mesure de l’énergie cinétique des photoélectrons éjectés des atomes par les rayons X. L’XPS apporte cependant généralement une information « intégrée » sur une certaine profondeur, sans accès en détail à la répartition des différentes espèces à l’interface.
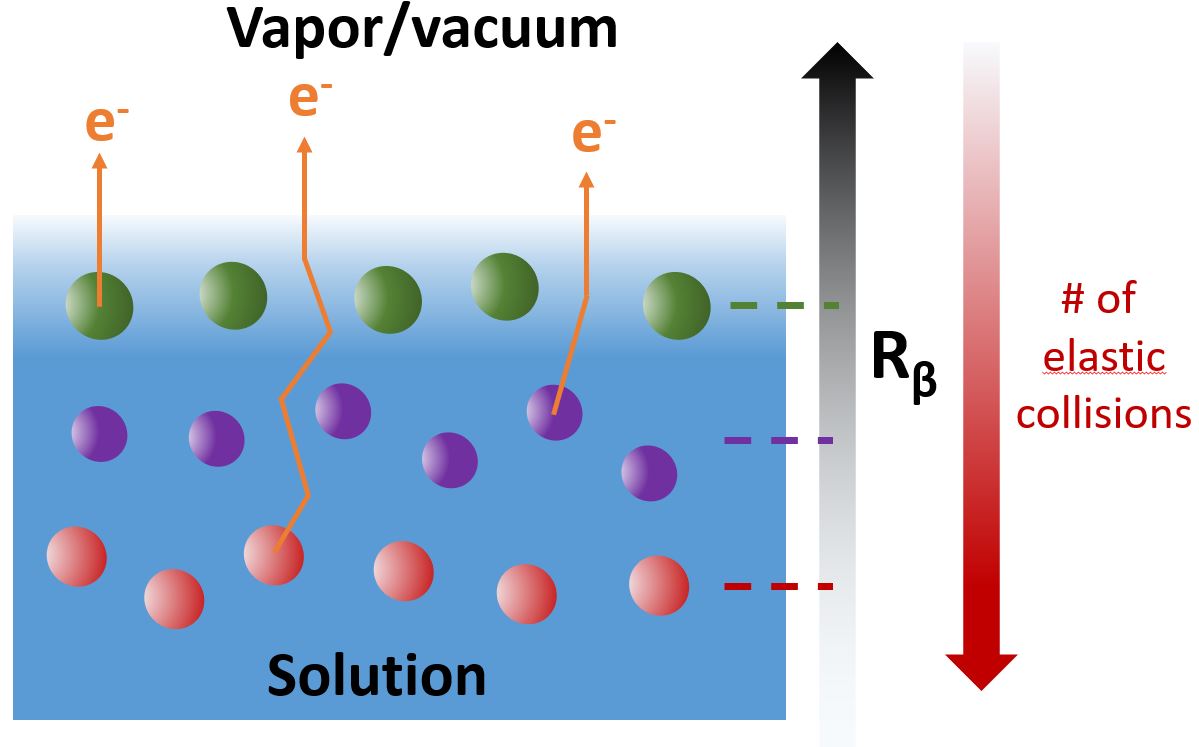
C’est une variante de l’XPS, qui consiste à mesurer aussi l’angle d’émission des photoélectrons, qui a été utilisée ici. Dans le liquide, les électrons arrachés aux atomes par les rayons X ne suivent pas forcément une trajectoire rectiligne : ils peuvent être déviés par des collisions élastiques avec les molécules environnantes. On peut quantifier à quel point ils sont déviés en moyenne en comparant la distribution angulaire globale des électrons pour une molécule isolée, en phase gazeuse (où tous les électrons suivent des trajectoires rectilignes), et la même molécule en phase liquide. Or, plus un atome se situe profondément au sein du liquide, plus les électrons qui en sont éjectés subiront de collisions élastiques avant de sortir. La quantité de déviation de la distribution angulaire devient alors un indicateur de la profondeur moyenne de l’atome sondé : c’est le principe schématisé sur la figure 1. La structure en « couches » (irréaliste mais illustrative) des différentes espèces schématisées dans la figure serait difficile à déduire de mesures XPS normales mais devrait immédiatement ressortir de la mesure des distributions angulaires.
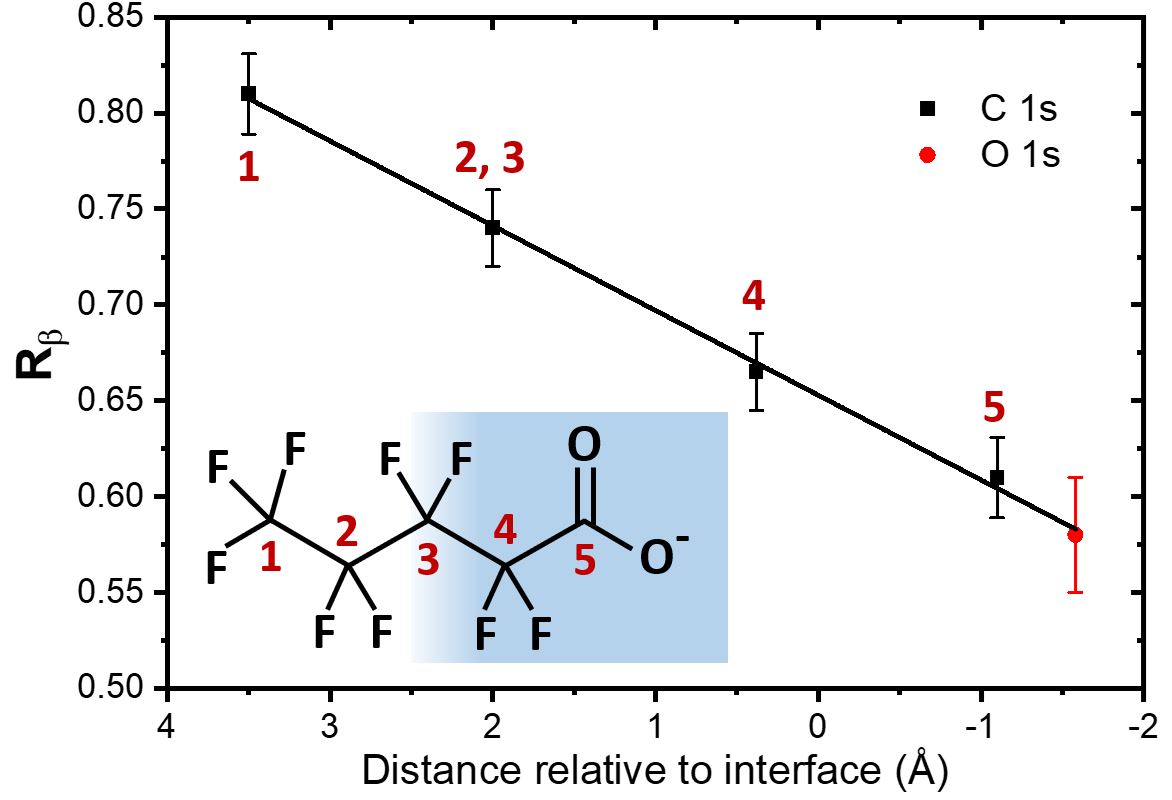
L’équipe a démontré la validité de ce principe en utilisant une molécule modèle de surfactant, c’est-à-dire une molécule ayant tendance à se placer à l’interface entre liquide et gaz, avec une partie hydrophobe en dehors du liquide et une partie hydrophile à l’intérieur (cf. insert de la figure 2). Il est possible de distinguer par XPS quatre des cinq atomes de carbone présents dans la molécule (car ils ont des environnements chimiques suffisamment différents). Ces quatre groupes d’atomes de carbone se distribuent le long de l’interface à différentes profondeurs, comme l’ont également montré précisément des simulations réalisées par des théoriciens impliqués dans cette étude. Comme montré sur la figure 2, chaque groupe d’atomes présente une déviation entre la distribution angulaire gaz et liquide (quantifiée par un facteur Rβ non détaillé ici) différente, alors même que ces atomes ne sont séparés que d’environ 1,5 Å, soit 0,15 milliardième de mètre.
Ces mesures démontrent tout le potentiel de cette technique pour déterminer des profils en profondeur à l’interface liquide-vapeur – mais également potentiellement sur d’autres types de surfaces amorphes, solides par exemple.