Hidden pitfalls of X-ray photoelectron spectroscopy for quantitative analysis
This work was performed in an international collaboration between nearly 20 researchers, including both experimentalist and theoreticians, from 6 countries: France, Sweden, Norway, Finland, US and Italy, and is a result of almost 40 years of observations and research since the early days of X-ray photoelectron spectroscopy (XPS). The aim is to determine the intrinsic limitations to a quantitative interpretation of spectral signals in this technique.
XPS, also known as photoemission spectroscopy (PES) or electron spectroscopy for chemical analysis (ESCA), is a widely used analytical technique that can be applied to a broad range of materials for characterization and for obtaining valuable quantitative and chemical-state information. This technique is based on Einstein’s photoelectric law for emission of electrons, so-called photoelectrons, upon irradiation of the material with photons. The discovery of this law brought Einstein the Nobel prize in 1921. Noteworthy, Manne Siegbahn was also awarded the Nobel Prize in 1924 for the development of X-ray spectroscopy and later, in 1981, his son Kai Siegbahn received the Nobel Prize for the development of the ESCA technique.
The benefits of XPS technique, most notably both element and chemical-state specificity, were rapidly recognized and highly appreciated not only in scientific community but also in industries. Provided that the energy of the photon source is tunable, it can be used in a non-destructive manner for depth profiling. This is due to the strong influence of the kinetic energy of photoelectrons on the depth from which they can escape from the material. Therefore, by varying the photon energy one can enhance or decrease the signal from the surface relative to the bulk. The same principle has been recently extended to the studies of liquids allowing, e.g. determination of concentrations and geometrical orientation of long-chain organic molecules on the surfaces of solutions, which can vary substantially from those of the bulk.
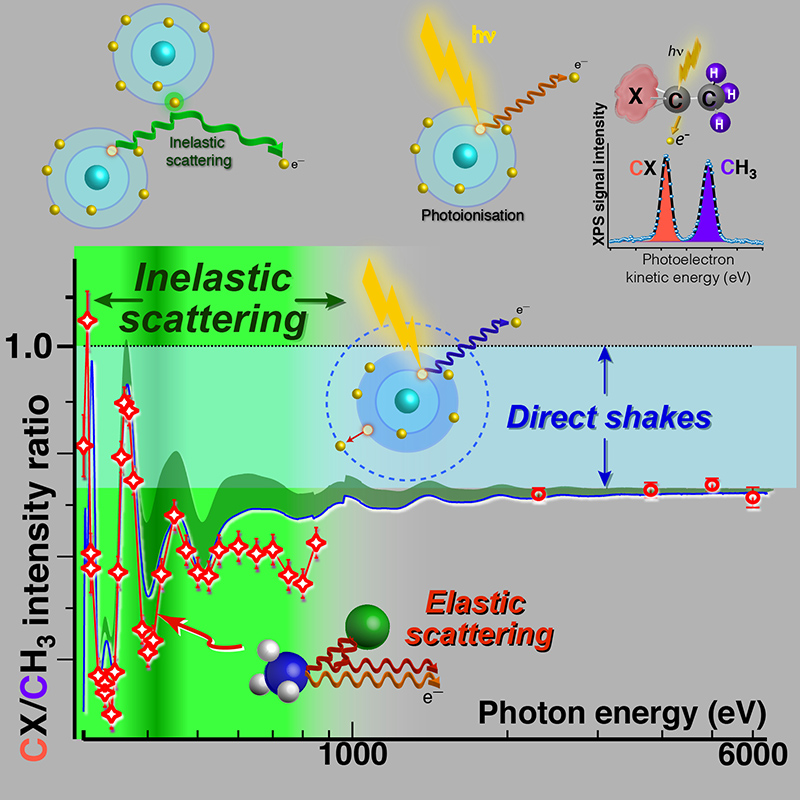
To perform such quantitative analysis one needs to know cross-sections (i.e. probabilities) for photoemission. Nowadays, theoretical cross-sections for atoms are most commonly used, with the implied assumption that the molecular environment does not play a significant role on the strength of the measured photoelectron signals. However, the present work shows that this assumption is not justified, especially for systems containing heavy elements and/or molecular groups with double or triple bonds.
As a first approximation, the intensity of the XPS signal should be proportional to the number of atoms of any specific type in the sample. However, already in the early days of XPS as a tool for qualitative and quantitative analysis, it was observed that measured photoelectron intensities might deviate (sometimes significantly) from the expected values. However, no systematic studies were performed to characterize these deviations and elucidate underlying phenomena.
This study was performed in a large international collaboration of nearly 20 researchers, counting both experimentalist and theoreticians, from 6 countries: France, Sweden, Norway, Finland, US and Italy. XPS spectra were recorded at the soft X-ray beamlines PLEIADES at SOLEIL and I411 at MAXII (Sweden) as well as at the tender X-ray beamline GALAXIES at SOLEIL.
The molecules were chosen so that all carbon atoms within a molecule (2 or more) have very different chemical environments and hence can be distinguished easily in photoelectron spectra allowing simple quantification of XPS signals for each of the carbon atoms. From the stoichiometry, intensity ratios for two separate carbon XPS peaks belonging to the same molecule are expected to be 1 (1:1) as there is only 1 carbon atom of each type in the molecule. However, experimental XPS ratios, measured over an extended energy range, not only deviated from the expected value of 1 but also showed pronounced oscillations resulting in changes of the XPS signal up to 40%. These intensity modulations are caused by scattering of the photoelectron inside the single molecule (similar to another spectroscopic technique, extended X-ray absorption fine structure EXAFS, or photoelectron diffraction), which is typically (and as shown in this work, erroneously) ignored for small isolated molecules. The intensity oscillations are found to be pronounced in the soft X-ray energy range for electron kinetic energies up to ~600 eV.
Noteworthy, even at high photon energies where oscillations due to elastic scattering are damped, the measured XPS ratios do not reach the expected stoichiometric value of 1 due to multi-electron processes always accompanying photoionization (e.g. direct shake-up and shake-off1 processes).
Moreover, in this work it was found that there is one additional important source for intensity losses even in small isolated molecules, which is due to internal inelastic scattering and is the strongest when kinetic energy of the escaping photoelectron is low. On one hand, the observation of internal inelastic scattering, related to the so-called inelastic mean free path (IMFP), in a single small molecule was unexpected. So far, this effect has been considered only for solid/liquid samples, while its importance was assumed negligible for dilute gas phase systems. On the other hand, the observation of inelastic intramolecular scattering in an isolated system lies at the origin of IMFP and surface sensitivity of XPS.
Any scientist or specialist using XPS for qualitative and quantitative sample analysis should consider the conclusions of this work to avoid erroneous interpretations of their data. The method suggested in this work for separation between the dominant effects accompanying photoionization could be used to develop more accurate theoretical models that, in turn, would facilitate more accurate data analysis.
1 - During a photoionization event, the ejection of an electron (due to the electromagnetic radiation) from a deep layer of the considered atom can trigger the ejection of another electron from the same atom (shake-off) or its transition to a higher layer (shake-up).