Dye-sensitised solar cells - How fast is ultra-fast charge transfer?
Electron transfer between a molecule and a semiconductor surface is a central mechanism in devices such as dye-sensitised photovoltaics and water splitting photoelectrochemical cells. A collaboration led by the University of Nottingham between the SEXTANTS beamline scientists and researchers at Nottingham and Lund Universities has explored this process on the low femtosecond (fs, i.e. 10-15s) timescale using resonant inelastic x-ray scattering (RIXS).
In a dye-sensitised solar cell – either for generating electrical power or splitting water - a light-harvesting molecule first absorbs a photon of visible light through the excitation of an electron from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). This excited electron is then transferred into the empty conduction band of a wide bandgap semiconductor surface to which it is coupled and carried away through the rest of the device to make use of the harvested energy.
The electron transfer process from the molecule to the oxide lies at the heart of these devices and in particular the efficiency of this charge transfer process. In this study, the molecule in question is bi-isonicotinic acid, the anchoring ligand or many of the most efficient organometallic dye molecules, and the surface is a single crystal of rutile titanium dioxide, TiO2(110).
In a working solar cell, excitation to the LUMO is induced by a photon of visible light. In this experiment, a soft x-ray photon (of higher energy) is used instead to excite an electron from the N1s core level of the nitrogen atoms in the molecule into the LUMO (and other higher lying unoccupied molecular orbitals). Now the electron can participate in two competing processes. The first is charge transfer into the oxide just like in the solar cell. The second is to participate in a transition that refills the core hole. This competing process is very fast – the lifetime of the core hole is only around 6 fs. By identifying the spectroscopic signature of this core hole decay process in the coupled system we can see which process is faster: core hole decay or charge transfer – and we can also explore other processes that happen in this short-lived core-excited state.
In this study, the molecules were measured in two different regimes: decoupled from the oxide in a thick multilayer film and electronically coupled to the surface in a chemically bound monolayer. Using the multilayer, where the excited electrons have a sufficiently long lifetime to participate in the core hole decay, the scientists identified the ‘participator’ channel in the N1s RIXS around the elastic scattering energy. They also discovered strong vibronic features within this channel where the excited electron can couple to the lowest vibrational state of the molecule even on the ultra-fast timescale of the core hole lifetime.
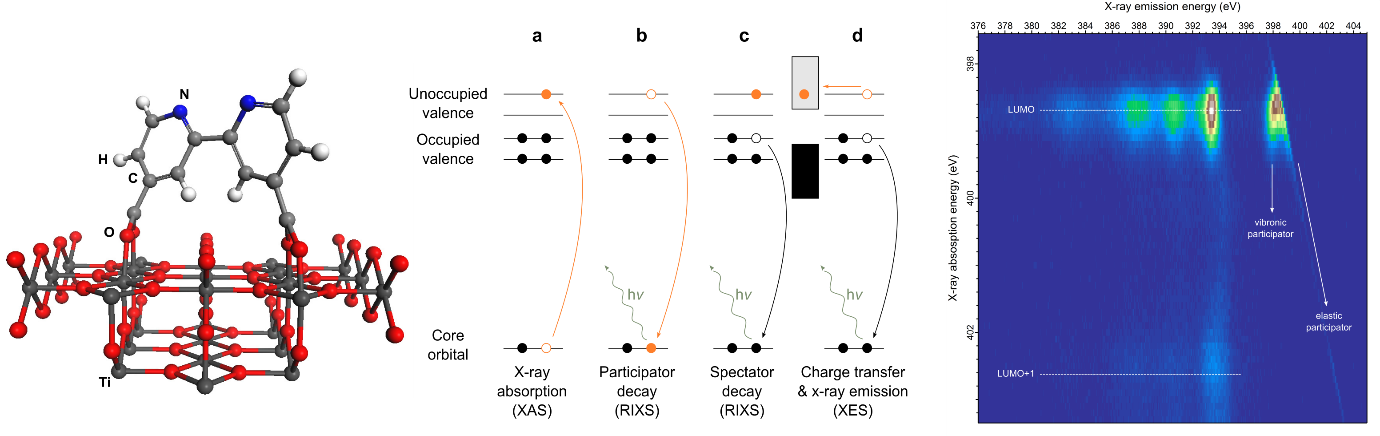
Using the monolayer, where the excited electrons in LUMO-states that lie above the conduction band edge of the underlying oxide are known to have very short lifetimes on the order of 3 fs, the team were able to explore the effect of ultra-fast charge transfer on the ‘participator’ channel in the RIXS. This is possible because the LUMO lies below the conduction band edge so there are no empty states to tunnel into, offering a direct comparison between states where charge transfer is forbidden and states with charge transfer takes place on the timescale of the core-hole lifetime. Interestingly, the participator channel and vibronic coupling were not significantly depleted by ultra-fast charge transfer of the excited electrons to the oxide conduction band suggesting a gap in our understanding of how these processes affect RIXS compared to the related non-radiative technique of RPES (resonant photoelectron spectroscopy).
This article has been selected as Journal of Chemical Physics 2017 Editors’ Choice article.