Cellular regulation. VHL E3 ubiquitin ligase, a new potential druggable protein
Together with protein phosphorylation, protein ubiquitination plays one of the most important role in cellular regulation. Defects in both signaling pathways are involved in cancer and other disease mechanisms. The research described here, which is the result of the collaboration between scientists at Cambridge and Yale Universities, demonstrates for the first time that one of the enzymes involved in the protein ubiquitination is a druggable protein.
Protein ubiquitination1 occurs through a cascade of enzymatic reactions, involving an E1 ubiquitin-activating enzyme, an E2 ubiquitin-conjugating enzyme and an E3 ubiquitin ligase. E3 ubiquitin ligases confer substrate specificity to protein ubiquitination pathways, and are thus attractive drug targets. However, to date, efforts to target E3 ligases using small molecules have been rewarded with limited success, resulting in this protein family being considered as “undruggable”.
The VHL/ Hif-1α transcription factor interaction, involved in several pathologies
The research described here demonstrates for the first time that the von Hippel-Lindau (VHL) E3 ubiquitin ligase is a druggable protein. This multi-subunit E3 complex, which comprises of pVHL, ElonginB, ElonginC, Cullin2 and Rbx1 proteins, plays a major role in cellular oxygen sensing by catalyzing the poly-ubiquitination and subsequent degradation of the transcription factor Hypoxia Inducible Factor 1 (Hif-1a). Mutations in pVHL that weaken or disrupt the pVHL:Hif-1a interaction, or perturb Hif-1a ubiquitination, give rise to VHL disease, an inherited human cancer syndrome with a high occurrence of retinal, spinal and cerebellar haemangioblastomata, clear cell renal carcinoma, and phaeochromocytoma. This supports the correlation between upregulation of Hif-1a and tumor development, and the role of the VHL E3 ligase as a tumour suppressor (1).
On the other hand, the activation of this protein has a positive impact on pathologies such as chronic anaemia or strokes. It is thus a key-protein, which activation or, on the contrary, degradation, plays a role in various pathologies.
Several methods to design inhibitors
In ref.2 is described the discovery of the first micromolar-affinity inhibitors for the pVHL:Hif-1a interaction. The compounds were rationally designed using as a starting point the key hydroxyproline (Hyp) residue that, as part of Hif-1α, is crucial for binding to pVHL (3-5). Molecules susceptible to link this site constitute particularly interesting ligands: they could afterward be used as PROTACS (Proteolysis Targeting Chimeric Molecules), in order to target a protein of interest for degradation by ubiquitinylation (one part of the PROTAC is a binding site to VHL, another part links the target protein to be destroyed). Using de novo computational tools in silico, we designed promising Hyp analogues that would be elaborated to occupy the left-hand side (LHS) and right-hand side (RHS) of this protein-protein interaction (PPI) (Figure 1a and 1b). Compound 1, bearing an isoxazole group at the LHS, proved a promising pharmacophore to interact with a conserved water molecule. Further elaboration of the RHS guided by our crystal structures of pVHL with the soaked ligands bound, led to the optimization of 2 which exhibited affinities down to single-digit micromolar as determined by isothermal titration calorimetry (ITC) and fluorescence polarization (Figure 1b).
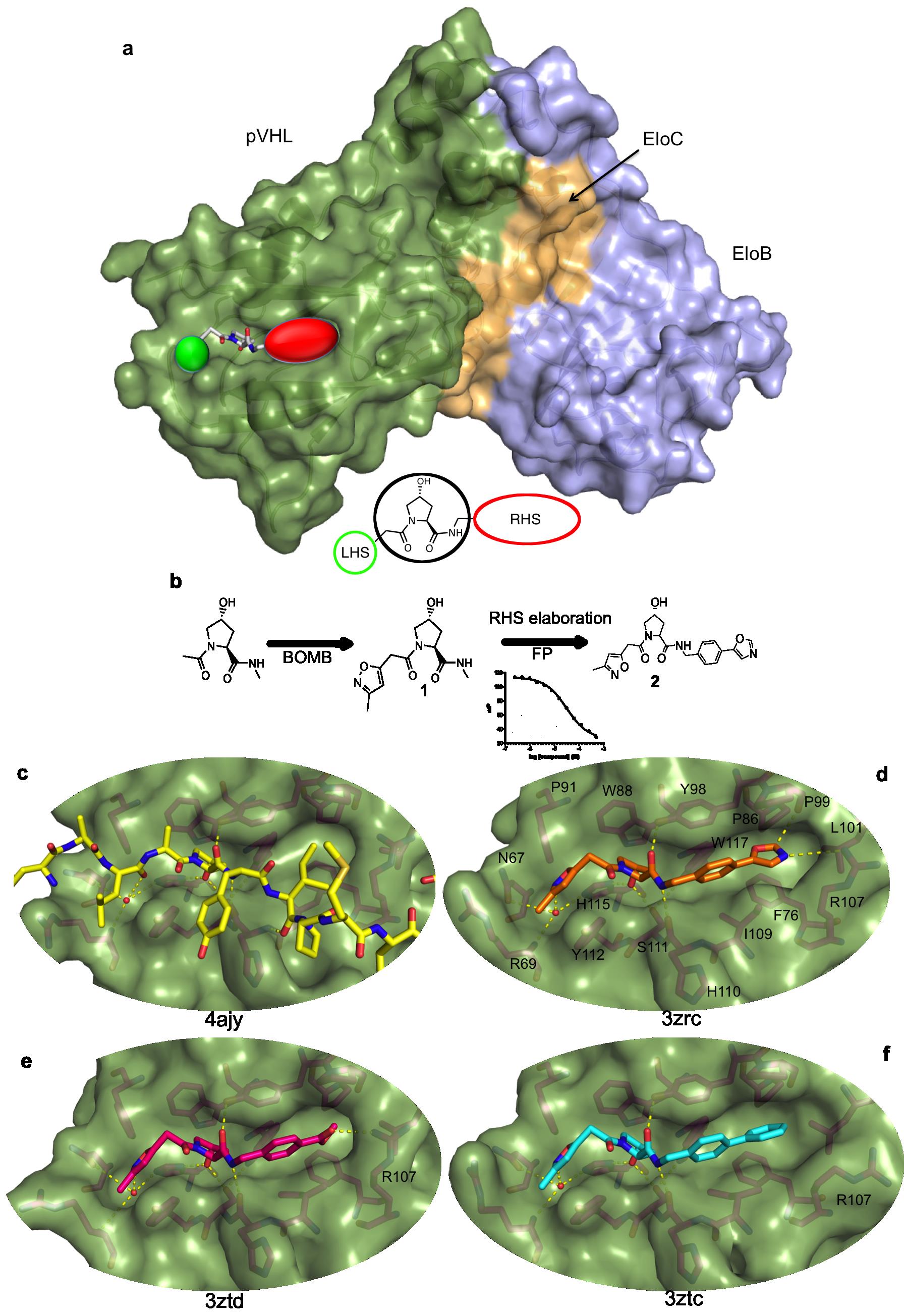
The X-ray structures of pVHL with bound 2, as well as a methylbenzoate and a biphenyl RHS analogue, show that the ligands mimic well the binding mode of the Hif-1a peptide (Figure 1c-f). They recapitulate the hydrogen bonding network around the Hyp core residue, and form a hydrogen bond with the water molecule at the LHS pocket, as hypothesized. Furthermore, the crystal structures point to a degree of conformational plasticity of the RHS pocket, with the bulkier biphenyl group inducing a significant movement of the Arg107 side chain (Figure 1f) and the optimal oxazole group forming hydrogen bonds with the Arg107 NH2 and with the Pro99 backbone carbonyl (Figure 1d).
Limits of the conventional screening methods
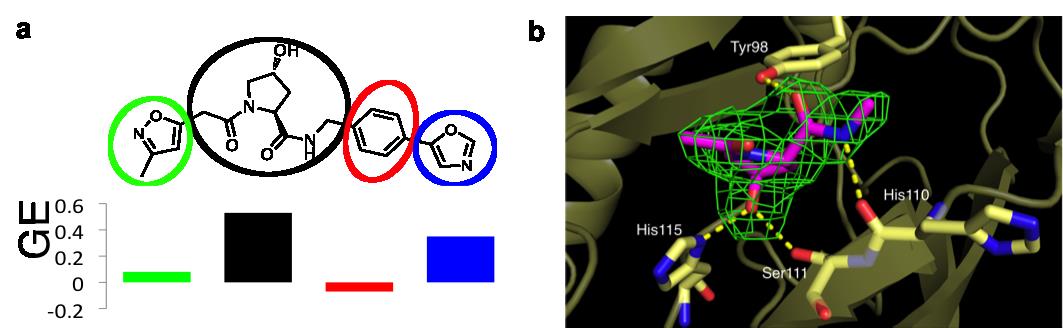
This discovery prompted the authors to ask whether they could have found these compounds in a fragment-based screening approach. Starting from the three ligands shown in Figure 1, they modularly “decomposed“ them into smaller and smaller fragments that should in principle target the LHS, the Hyp core and the RHS at this PPI. The results of this study are described in ref.6. Using biophysical techniques commonly employed for fragment screening, they could only detect binding of fragments that are more elaborate than those typically screened against classical “druggable” targets, and that violate several widely-accepted guidelines that are used to assemble fragment libraries. Furthermore, they dissected the individual contributions of different groups to the ligand binding free energy by applying specific “efficiency” metrics that take into account binding affinities, size and lipophilicity. Despite its modest affinity that escaped its detection in biophysical screens, the Hyp core was found to contribute the most to the ligand binding free energy (Figure 2a) and to recapitulate the expected interactions as observed in a crystal structure of the Hyp fragment bound to pVHL (Figure 2b).
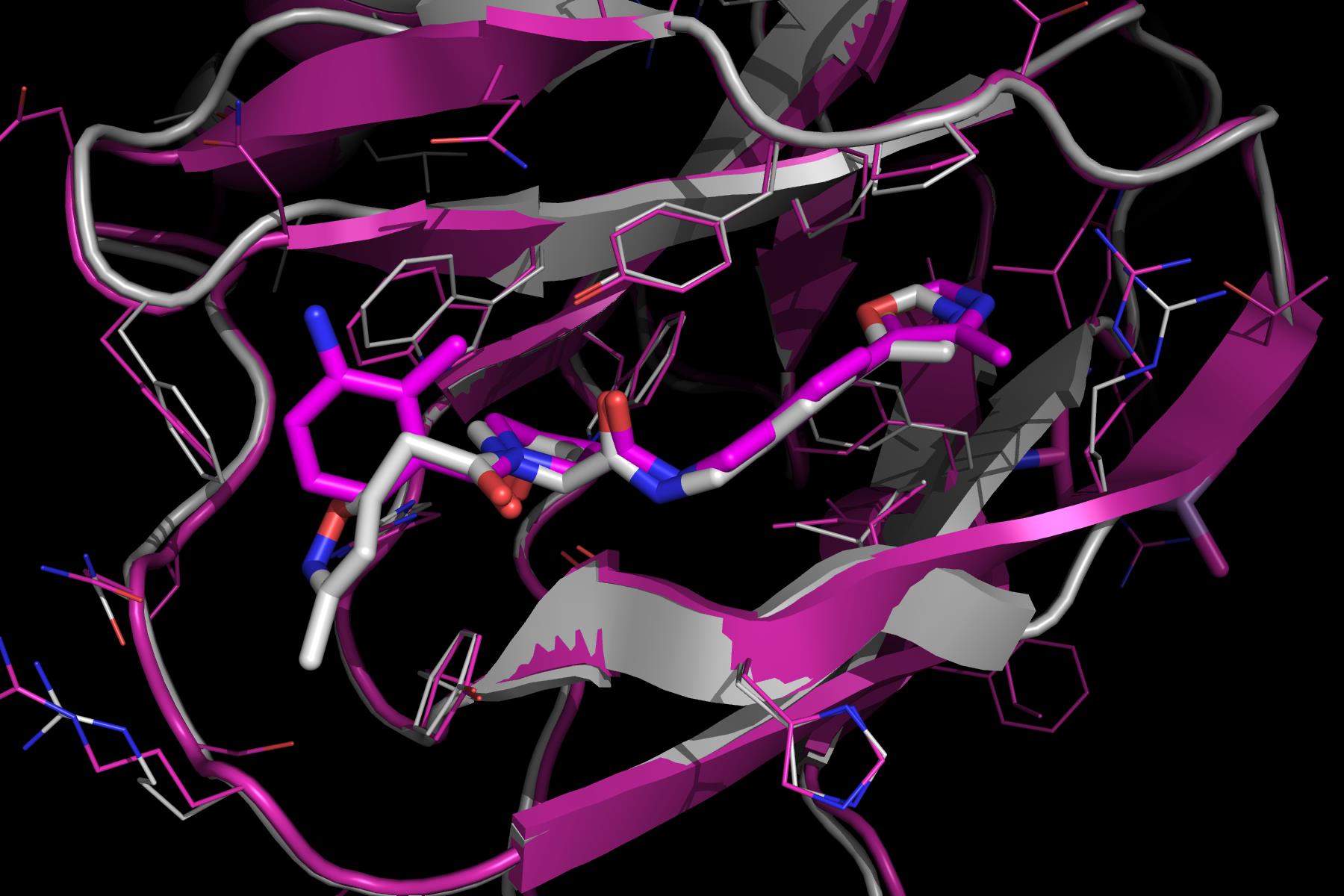
In a subsequent study, described in ref. 7, further medicinal chemistry efforts around the LHS and RHS groups led to the development of the first sub-micromolar inhibitors. At first instance, the RHS oxazole moiety of 2 was optimized to a 4-methylthiazole. Subsequently, the LHS moiety was also optimized, ultimately leading to incorporation of a 3-amino-2-methylbenzamide group, and to a Kd of 0.9 µM. The crystal structure of the final compound (3) shows that the RHS methylthiazol binds in a similar conformation to the oxazole group of 2 (Figure 3). Interestingly, the LHS group clearly binds to pVHL in an alternative fashion. While the isoxazole group of 2 interacted with the structural water as shown above (Figure 1d), the benzamide instead is oriented away from the water pocket, and lies adjacent to the side chain of Trp88. The aniline appears to make a completely novel water-mediated H-bond to the side chain of Gln96.
Conclusion
The structure-based design efforts have lead to the discovery of the first potent class of small molecules targeting the VHL E3 ubiquitin ligase and disrupting its interaction with HIF-1a, thereby demonstrating the “druggability” of this challenging PPI. Such an integrated approach combining structural biology with medicinal chemistry may prove useful for targeting PPIs of other E3 ligases.
Finally, and perhaps more importantly, these results serve as a cautionary tale for drug hunters interested on targeting PPIs using fragment-based approaches, warning that excellent chemical starting points (such as Hyp) could be well overlooked/missed from standard screens, as they could bind too weakly to be detected using routine methods and/or they could be filtered out of conventional fragment libraries.
1 – Ubiquitination: Ubiquitin is a small protein present in all the subcellular compartments of the eukaryotic cells (it is ubiquitous). Its role- ubiquitination -is to label other proteins in order to direct them to compartments in the cell, including the proteasome which destroys and recycles proteins. For that purpose, several molecules of ubiquitin are covalently linked to the target protein, thanks to the action of three enzymes: E1, E2 and E3-ligases.
2- hemangioblastomata: Vascular tumor of the nervous system.
3 - pheochromocytoma: Tumor of the adrenal glands
4 – pharmacophore: Molecule feature which is necessary for the biological activity of this molecule.
References
(1) Kaelin, W.G. Nat. Rev. Cancer 2002 2, 673
(2) Buckley, D.L. et al. J. Am. Chem. Soc. 2012 134, 4465–4468
(3) Hon, W-C. et al. Nature 2002 417, 975
(4) Min, J-H. et al. Science 2002 296, 1886
(5) Loenarz, C. et al. Angew. Chem. Int. Ed. 2009 48, 1784
(6) Van Molle, I. et al. Chemistry & Biology 2012 19, 1300
(7) Buckley, D. L. et al. Angew. Chem. Int. Ed. Engl. 2012 51, 11463