Transmission of nerve impulses – New data about the 3D structure of ion channels toward a better understanding of mechanism for ion permeation
The pentameric ligand-gated ion channels (pLGICs) constitute a large family of ionotropic receptors that are ubiquitously represented in the animal kingdom. In vertebrates, the family encompasses cation selective serotonin receptors and nicotinic acetylcholine receptors on one-hand, and anion selective GABA and glycine receptors on the other hand. Read also: http://www.synchrotron-soleil.fr/Soleil/ToutesActualites/2013/PX1-ELIC-PNAS.
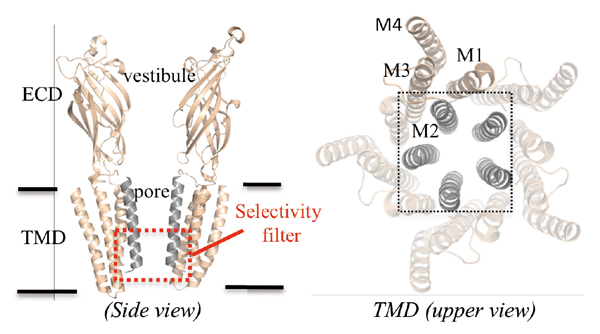
The pLGICs couple agonist binding at the extracellular domain to the opening of their transmembrane domain composed of four α-helices (named M1 to M4). All these receptors share a conserved molecular architecture based upon a five-fold symmetry with ions permeating through a central transmembrane pore bordered by the M2 helices.
Little is known about the molecular mechanisms underpinning permeation and charge selectivity (Fig. 1). The crystal structure of GLIC, a prokaryotic homolog of the family was solved at 2.9 Å resolution in an open form of the channel and provided a significant advance in the possibility to study ion permeation in pLGICs. The pore of GLIC is bordered by the M2 helices which successively expose six rings of residues to the lumen, from the top to the bottom, one ring of Glu243, three rings of hydrophobic residues Ile240, Ala237, Ile233, two rings of polar residues Ser230, Thr226 and finally Glu222. The pore is funnel-shaped, with a large upper diameter of 11Å and a constriction of 5Å diameter at the 226 and 222’ level that hosts the charge selectivity filter.
Combining X-ray crystallography, electrophysiology, MD simulations and electrostatic calculations provides novel insights on the molecular mechanisms of ion permeation in pLGICs, which are complex mechanisms as they involve protein residues, ions and water molecules that interact together dynamically and transiently as the ions flow down the channel.
The hydration in the pore of a ligand-gated ion channel is revealed with an unprecedented achieved level of details
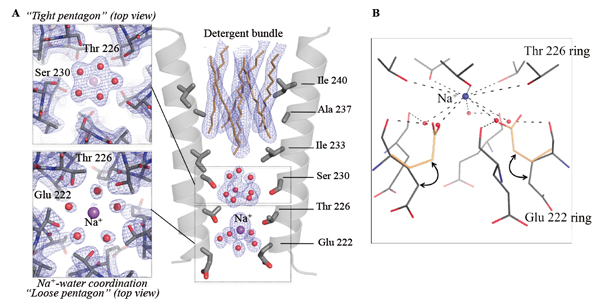
Understanding the molecular mechanisms of ion permeation in pLGICs requires documenting the interactions between ions, waters and the side chains of the M2 pore-lining residues that are thought to be crucial for ion transport. This kind of description is difficult to derive from existing pLGIC structures given the limited structural resolution achieved previously by X-ray crystallography. To understand the molecular mechanism of ion permeation in pLGIC, we solved the structure of GLIC at 2.4Å, the highest resolution achieved on this family of protein. These data that were collected at the Proxima 1 beamline of the SOLEIL synchrotron reveal for the first time the hydration geometry in the pore of a pLGIC (Fig. 2).
This reveals ordered water molecules at the level of two rings of hydroxylated residues (named Ser 230 and Thr 226) that contribute to the ion selectivity filter. Two water pentagons are observed, a self-stabilized ice-like water pentagon and a second wider water pentagon, with one sodium ion between them. The presence of a Na+ ion at this location has been confirmed by substituting Na+ with cations that display anomalous dispersion properties (see below). The second pentagon is directly involved in the hydration shell of the Na+ ion. These water molecules interact both with the Na+ ion and the main-chain carbonyl oxygen atom of Glu-2’. In the GLIC structure, the Glu-2' carboxylate groups display significantly higher B-factors than the other residues of M2, suggesting that these residues are flexible and may adopt alternative conformations. The ε-O carboxylate atoms of the Glu-2’ residues probably continuously exchange their positions with individual water molecules of the sodium ion-bound water pentagon, thus facilitating ion transport.
Mapping the solvent molecules binding sites
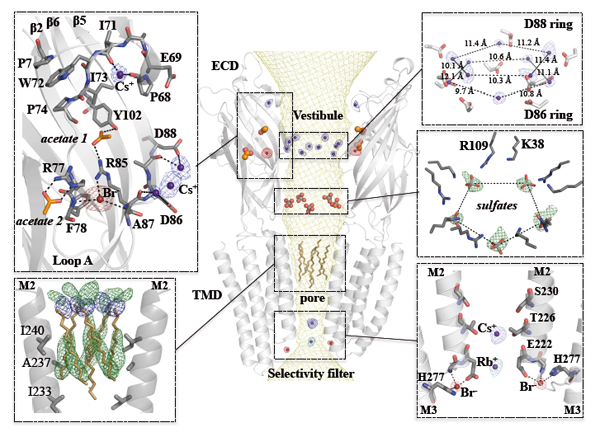
In order to fully characterize the functional implications of the organized water molecules observed in the pore and their role in ion permeation, it is necessary to identify the monovalent ion binding sites in the GLIC structure. To this aim, the protein was co-crystallized with Br-, Cs+, or Rb+ and X-ray data were collected at optimized wavelengths for each of these derivatives so as to maximize the anomalous signal (Fig. 3). These experiments were performed on Proxima-1 beamline taking advantage of the tuneable wavelength of the beamline. Peaks with an anomalous signal higher than 4σ were used to identify ion binding sites. The identified ion binding sites are essentially clustered in two distinct regions: in the ECD around the vestibule edge of the channel and in the TMD near the cytoplasmic end of the pore. The ion binding sites identified in the ECD are important for understanding the gating process, while the ion binding sites identified in the pore are important for understanding the permeation mechanism. Overall, these results suggest that permeating ions occupy at least two preferential positions in the most constricted region of the pore that has been shown to be the selectivity filter of these channels.
Toward a mechanism for ion permeation in ligand-gated ion channels
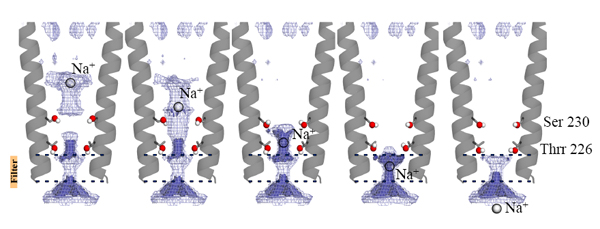
In order to further explore the roles of these water molecules and ion binding sites, we mutated important pore-lining residues and tested them by single-channel electrophysiology. Simulations and electrostatics calculations complemented the description of hydration in the pore. In particular, simulations that pull a cation through the pore reveal that the water pentagons observed in the crystal actively contribute to ion translocation when the ion is going through the pore. These water molecules are important for the ion to cross hydrophobic constriction barriers. Due to the strong sequence conservation of M2 pore-lining residues within the pLGIC family the conclusions presented in this study might be transferable to all the pLGIC channels (Fig. 4).
1 - Récepteur ionotrope : protéine membranaire interagissant avec un ligand, dont la fixation provoque l’ouverture d’un canal dans la protéine. Ce canal permet alors la traversée de la membrane par des ions.
2 – Agoniste : se dit d’un ligand dont la fixation à un récepteur provoque un changement de conformation de ce dernier, ce qui induit une réponse moléculaire (ici : traversée du canal par les ions).