First Structure of Pathogenic protein Huntingtin and its Link with Huntington’s Disease
Huntington’s disease (HD) is a deadly neurodegenerative pathology that develops in individuals that have more than 35 consecutive glutamine amino acids at one extremity of a protein called huntingtin. Which are the changes experienced by huntingtin when this pathological threshold is surpassed, causing the disease? By combining a set of biophysical tools, scientists from Centre de Biologie Structurale (CBS, Montpellier) solved this protein structure: it consists of an equilibrium of long partially helical conformations. The stability of these helices defines the capacity of the protein to aggregate and form inclusions in cells, which are at the origin of HD.
Huntington’s Disease (HD) is a deadly neurodegenerative disorder caused by an expansion of part of the gene called HTT (for huntingtin). When this part of the gene (httex1) is translated, if it has such an expansion it results in a huntingtin protein containing a long poly-glutamine (poly-Q) amino acid sequence at the N-terminal extremity. Only people with a succession of more than 35 consecutive glutamines develop HD. The structural changes that occur in the httex1 part when its length exceeds this pathological threshold of 35 glutamines remain poorly understood, precluding a molecular understanding of the pathology. The high-resolution structural investigation of httex1 had been considered as impossible due to its intrinsic flexibility, which precludes the use of X-ray crystallography and cryo-Electron microscopy, and the repetition of the same amino acid that hampers the use of traditional NMR approaches.
During the last years, the Highly Flexible Proteins group leaded by Pau Bernadó at the Centre de Biologie Structurale (CBS) in Montpellier has developed novel chemical biology approaches to specifically label specific positions of proteins with "heavy" amino acids (incorporation into the protein of heavy - but non-radioactive - isotopes of certain atoms, such as 13C to replace 12C), enabling the high-resolution Nuclear Magnetic Resonance (NMR) investigation of proteins with long repetitive sequences, such as httex1.
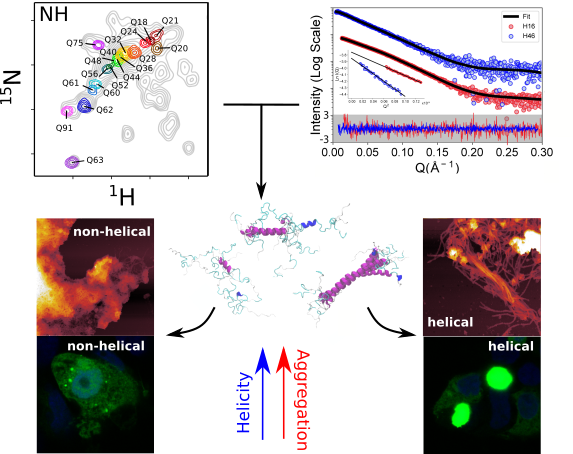
The systematic application of such a site-specific isotopic labelling has enabled the structural characterization of poly-Q tracts for pathogenic versions of httex1 with 46 and 66 consecutive glutamines. The information derived from NMR, which reports on the local structure, has been complemented with Small-Angle X-ray Scattering (SAXS) data measured at the SWING beamline, which reports on the global shape of the protein in solution.
The integration of these structural data in computational approaches developed at the LAAS laboratory in Toulouse by Juan Cortés yielded for the first time a structural model of a pathogenic version of huntingtin before aggregation.
The analysis made by the scientists reveals that the poly-Q tract adopts long α-helical conformations stabilized by glutamine side chain to backbone hydrogen bonds, and highlights the relevance of the residues flanking the poly-Q region in defining this structure. In collaboration with the group of Pierre-Emmanuel Milhiet (CBS-Montpellier), the scientists showed that α-helical stability is a stronger signature than the number of glutamines in defining aggregation kinetics, both in vitro and in cells, and the structure of the resulting fibrils as observed by Atomic Force Microscopy. Those observations provide a structural perspective of the pathogenicity of expanded httex1 in HD and pave the way to a deeper understanding of other diseases, such as Spinocerebellar ataxias, Spinal and bulbar muscular atrophy or Machado-Joseph disease, linked to the poly-Q expansion in certain human genes.