Combining computer design and structural biology to develop future leukemia drugs
Acute T-cell lymphoblastic leukemia is notorious for its high relapse rate and resistance to standard treatments, considerably limiting patients' chances of long-term survival. A team of researchers at the Centre de Recherche en Cancérologie de Marseille has developed a drug candidate by molecular design, based on the three-dimensional structure of an enzyme called deoxycytidine kinase, or dCK. In this study, this protein was targeted because of its recently identified role in the development of this type of cancer.
Acute T-cell lymphoblastic leukemia (ALL-T) frequently leads to cases of relapse and resistance after standard treatment, with a prognosis of only 5% survival at 5 years. This underlies the need for new therapeutic options for resistant patients. The uncontrolled proliferation of cancerous T lymphoid cells results in a high metabolic demand, necessitating the production of a large quantity of essential components for these cells. A main component is DNA, synthesized by two pathways: the main “de novo” synthesis pathway, and an alternative pathway known as "salvage", in which the enzyme deoxycytidine kinase (dCK) plays a pivotal role. Thus, dCK has recently emerged as a target of interest in cancers for which the salvage pathway is essential.
Scientists from the research teams of Xavier Morelli and Patrice Dubreuil at the Centre de Recherche en Cancérologie de Marseille have discovered a potential drug that targets dCK using a strategy that seamlessly blends computational and experimental methods. They combine the design of active molecules through computational approaches utilizing the atomic structure of the target with organic chemistry syntheses and biological validations conducted in vitro, on cellular models and in mice.
In a previous study, the scientists had identified dCK as the secondary target of a drug called masitinib, already used in the treatment of certain tumors. Masitinib operates by inhibiting enzymes within the kinase family. In this new research, they hypothesized that an increased level of interaction between masitinib and dCK could trigger inhibition of the enzyme, subsequently affecting DNA synthesis and the proliferation of cancer cells.
They therefore applied a drug design approach called DOTS (Diversity-oriented target-focused synthesis) to masitinib. Its aim is to increase the complementarity and hence the strength of the interaction between the molecules developed and the biological target - in this case, dCK. During the compound development process, they analyzed over 1000 potential molecules, ultimately selecting, synthesizing, and evaluating 74 compounds.
They studied the atomic-scale interaction between the target dCK and the compounds by conducting X-ray diffraction measurements on crystals of the target protein complexed with candidate molecules. The knowledge acquired on the PROXIMA-1 and PROXIMA-2 beamlines enabled them to propose rational modifications to the compounds' chemical structures, enhancing their effectiveness. After several optimization cycles, they obtained a drug candidate called OR0642, 1000 times more potent than the initial molecule, masitinib.
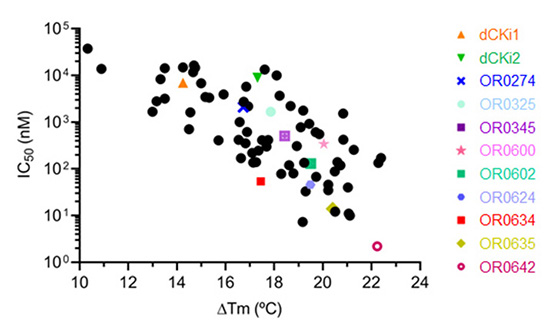
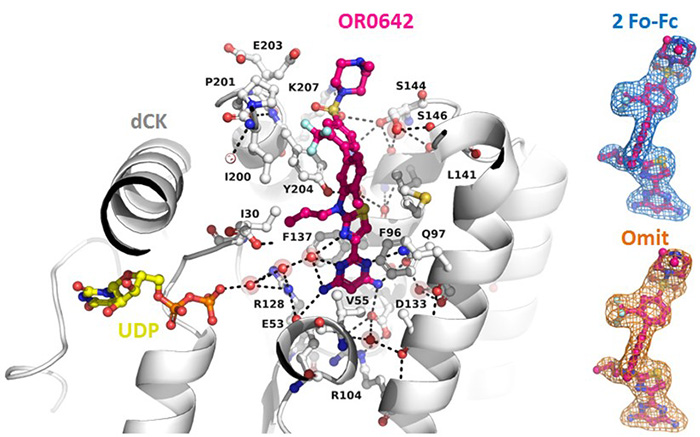
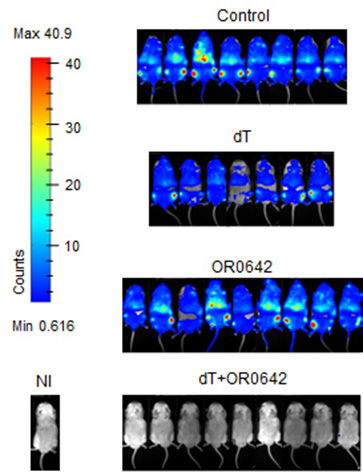
In combination with thymidine, a well-known physiological inhibitor of the de novo DNA synthesis pathway, the lead compound OR0642 inhibits dCK and the salvage DNA synthesis pathway. The effect on cellular models has been confirmed, and mice transplanted with tumor cells from ALL patients who have benefited from this therapy have doubled their survival rate. This therapy uses the concept of synthetic lethality, whereby only the combination of two molecules selectively targeting these two pathways is toxic to tumor cells. This result demonstrates the proof-of-concept of this computational molecular design strategy and its therapeutic potential in a particularly aggressive cancer.
Overall, this study demonstrates an innovative approach to drug design, where the repositioning of a drug (masitinib) on a new target (dCK) can be used as the starting point for a semi-automated drug design approach that could be applied to other targets, in cancer or for other pathologies. In addition, the research team is continuing to optimize ORO642 to transform this 'prototype' into a potential orally-administered drug, and also to find other applications in diseases where dCK plays an important role.
This work is published in the journal Nature Communications.