A new method to identify protein secondary structure and fold
Analysis of the structure of amyloid fibrils of various disease-related proteins, causing pathologies such as Alzheimer’s, Parkinson’s and Huntigton’s disease has been the focus of an international research effort between the Neuroimmunology Research Group of the Hungarian Academy of Sciences and Department of Biochemistry, Eötvös Loránd University, Budapest, Hungary, the Institute for Protein Research, Osaka University, Japan and the DISCO beamline at SOLEIL Synchrotron, France. Structural characterization of amyloid fibrils under physiological conditions down to the spatial arrangement of these proteins has been notoriously difficult. The international group has now reported that, with their novel algorithm and an enhanced spectroscopic approach using Synchrotron Radiation Circular Dichroism spectroscopy, the secondary structure determination of these protein formations became feasible.
Circular Dichroism (CD) spectroscopy is based on the differential absorption of left and right circularly polarized light. Biological chromophores, such as proteins have distinct CD spectra in the far ultraviolet region depending on their structure. Synchrotron Light Sources, renown for their increased light intensity and larger accessible spectral range as opposed to conventional light sources, have provided biophysicists with more accurate spectra accumulated in a shorter time and last but not least with an increased information content.
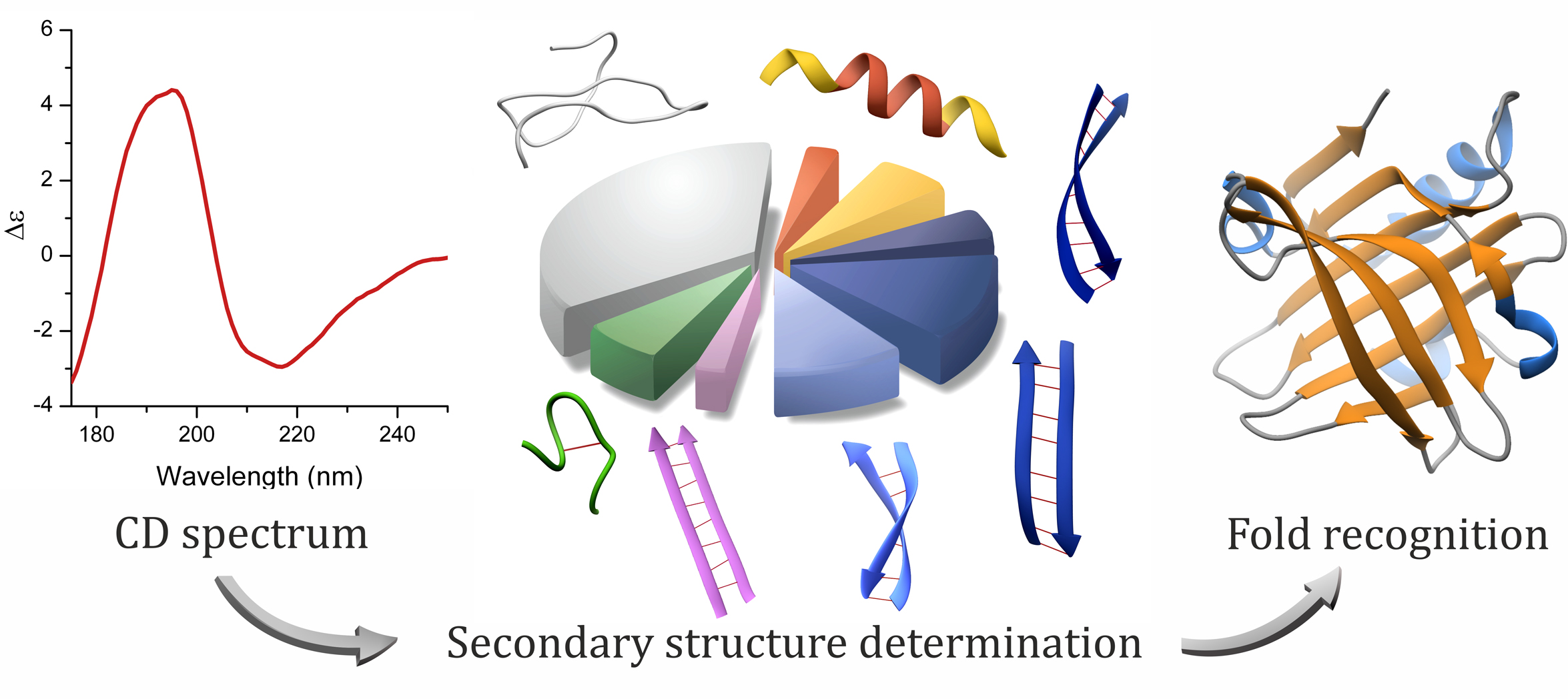
Careful analysis of the twisting of β-sheets, a secondary structure element in proteins, and their correlation with characteristic CD spectral features in the far UV range made a distinction possible between parallel and antiparallel β-sheets. The obtained increased structural information provided an accurate secondary structure determination and proved to allow the prediction of the protein fold. The protein fold is classified by the specific pattern of the secondary structure elements. The method is implemented in a novel bioinformatic algorithm (BeStSel). Besides β-sheet-rich structures, this method can be applied with good accuracy on any proteins. The researchers hope to help the community by providing it as a general tool for a quick and reliable structure analysis, which is especially useful when X-ray or Nuclear Magnetic Resonance techniques fail.