How solvation can affect the energy of the innermost electrons of an organic solute
Solvent molecules can easily alter the energy of the outermost electrons of an organic solute affecting the coloring of the solution, i. e. solvatochromism. Less obvious is the effect that solvation can have on the energy of the innermost electrons, the ones which are more tightly bound to the nuclei.
A recent study, performed on melamine in aqueous solution, revealed that the impact of the solvent on the inner electrons strongly depends on the nature of the chemical site where the electrons are located and if the site is involved in specific intermolecular interactions.
The energy of the inner electrons (commonly defined as binding energy, BE, the potential energy experienced by the electron in presence of a nucleus) is known to be the “fingerprint” of each element and of its chemical state (i.e. oxidation state). Hence, any variation in binding energy is indicative of a chemical change and can be exploited to shed light on many chemical processes. For instance, catalytic reactions are always more often investigated by means of X-ray Photoemission Spectroscopy (XPS). The technique, indeed, allows to measure the electrons binding energy of the catalyst -before, after and even during the reaction- with the possibility to identify the catalyst active sites and the reaction intermediates.
In this context, a team of scientists (University of Rome, University of Padova, Italy; Ruđer Bošković Institute, Croatia) chose to investigate how the electrons BEs of an organo-photocatalyst are affected by its interaction with water. The photocatalyst under investigation is the melamine molecule (triamino-s-triazine C3H6N6, see Figure 1), a compound that contains several nitrogen atoms, that is potentially suited to produce H2 from water under UV-visible light irradiation. According to theoretical investigations, upon photon-excitation, a proton-coupled electron transfer (PCET) occurs from the H-bonded water molecule to the C3N3 cycle, splitting the water in the OH radical and the H atom, the latter being bound to the triazine-N (N=C). An unambiguous way to identify such chemical species, and then to support the reaction mechanism, would be to monitor the BE variations of the N electrons in the melamine molecule before, during and after light illumination.
With the perspective to explore the overall reaction pathway, the research team exploited the XPS m-liquid jet technique available at the PLEIADES beamline to get the binding energy fingerprint of the nitrogen electrons in the melamine molecule, in the pre-reaction condition, i.e. in aqueous solution.
In order to rationalize the experimental findings, the scientists performed a two-step computational study. Firstly, classical metadynamics simulations were used to explore how melamine self-associate in aqueous solution and to identify a few basic dimeric configurations adopted by the molecule: by stacking (π-π interactions) and/or H-bonding interactions.
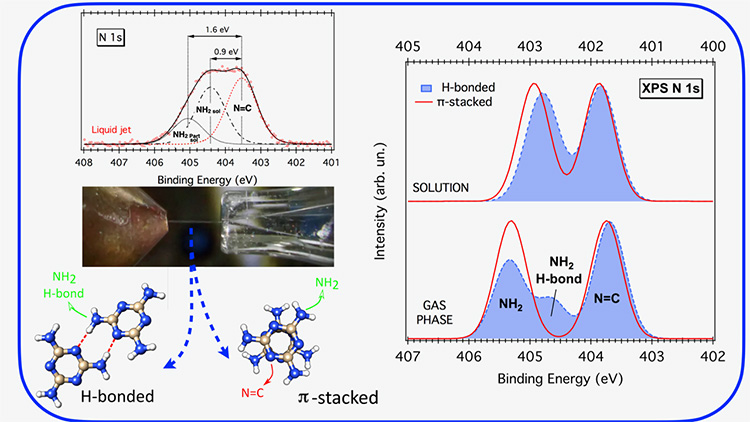
Within the second approach, XPS spectra of the melamine N electrons were computed for both the gas-phase and solvated dimers previously identified. In this way, the effect of solvation on each type of N-functional group (-NH2, N=C) could be analyzed. Pure π-stacked dimers show spectra almost identical to that of the monomer, where the chemical shift between the amino-N (NH2) and the triazine-N (N=C) energy levels notably decreases from ∼1.5 eV in the gas phase to ∼1.0 eV in solution (right-hand side of Figure 1). A more detailed analysis reveals that upon solvation, both energy levels shift to lower BEs, but the amino-N level shifts significantly more with respect to the triazine-N one (left-side hand of Figure 2).
Regarding H-bonded structures, the gas-phase spectra are sensibly different from those of pure π-stacked dimers (right-hand side of Figure 1). Surprisingly, the solvation suppresses all of the non-equivalences due to the H-bonds yielding similar XPS spectra for all dimers (right-hand side of Figure 1/Figure 2), matching very well the experimental spectra.
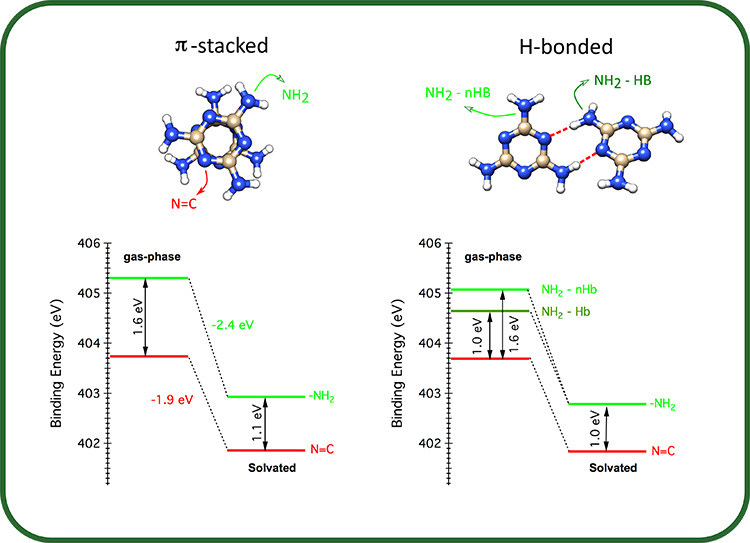
The results of this work show that solvation of melamine in water impacts more on the energy of the inner electrons located on the amino-N respect to those of the triazine-N. However, if the amino-N is involved in intermolecular H-bonding interactions, the impact of the solvent is reduced compared to the non-interacting configuration.